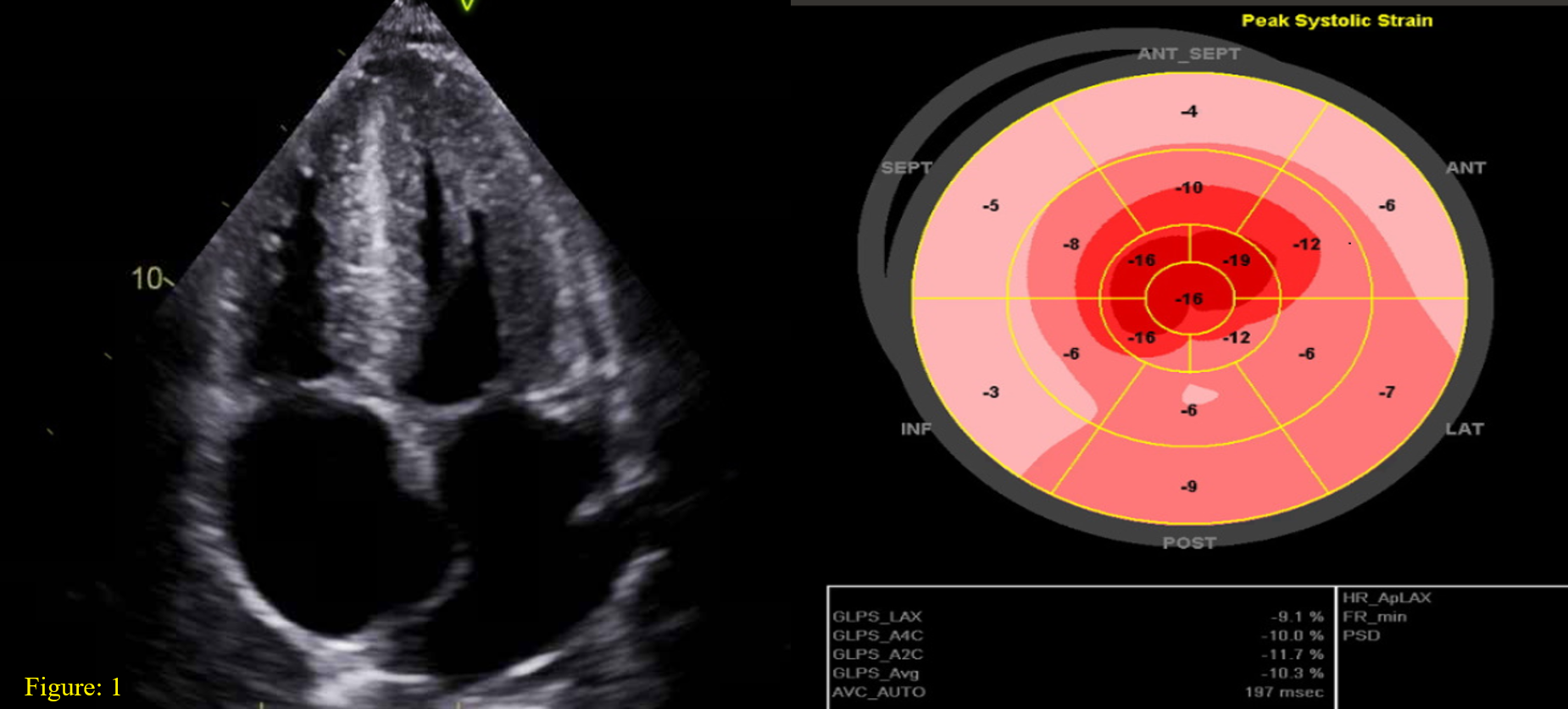
Case Presentation: Cardiac amyloidosis- a multiorgan disease, which carries a grave prognosis due to its rarity and nonspecific presentation. We report a case of systemic AL amyloidosis with predominant cardiac and renal involvement associated with multiple myeloma.• A 60-year-old male presented with progressively worsening anasarca including 3+ pitting edema in all extremities, and 80 lbs. weight gain for 8 months. • N-type pro-BNP and troponin T were elevated, and EKG showed diffuse low voltage QRS.• Echocardiography revealed findings suggestive of amyloidosis (Figure 1).• Light chain assessment showed a kappa to lambda ratio of 16.2. He had a new onset of acute kidney injury, requiring hemodialysis.• Endomyocardial biopsy revealed AL type cardiac amyloidosis (Figure 2) and bone marrow biopsy confirmed the diagnosis of Multiple Myeloma. • Patient received six cycles of bortezomib, cyclophosphamide, and dexamethasone. He suffered a PEA arrest with ROSC, but due to progressive deterioration, he was transitioned to comfort measures only.
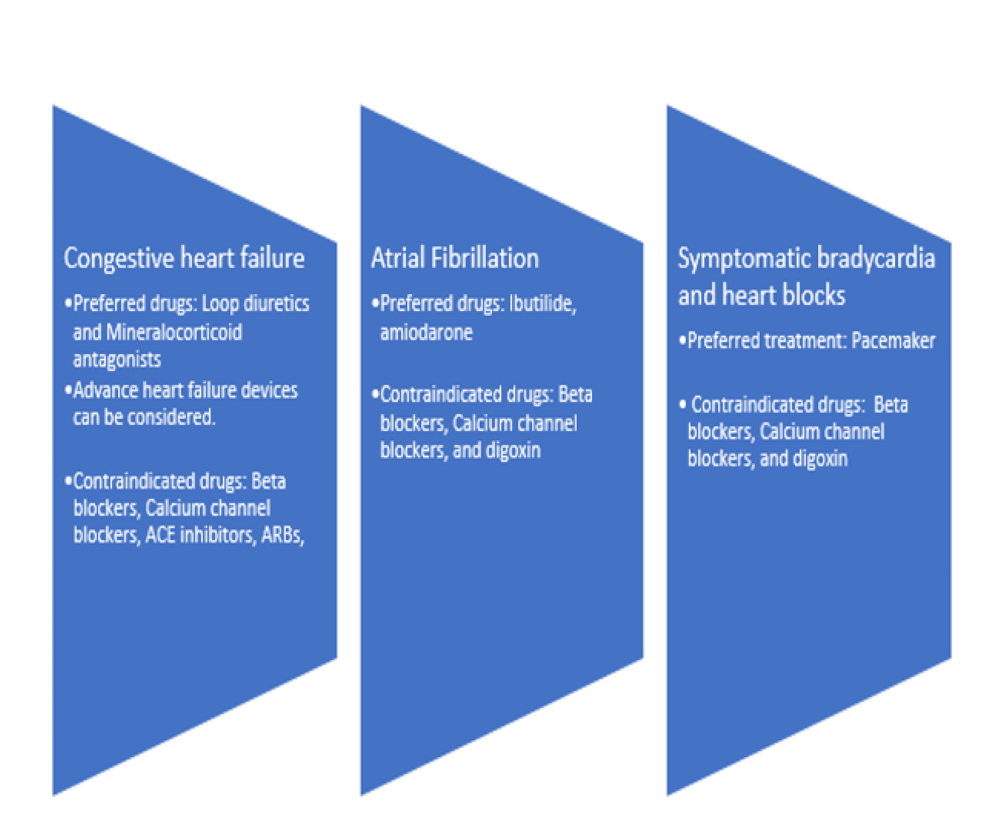
Discussion: • Cardiac involvement is seen ~60% of AL type amyloidosis associated with 6 months median survival [1].• Suspicion should instigate a thorough workup for underlying pathology driving amyloidosis such as plasma cell dyscrasia. • A low voltage EKG (QRS< 5 mm in all peripheral leads) is characteristic finding and echocardiogram findings outlined in Figure 1. • Cardiac magnetic resonance imaging can display late gadolinium enhancement secondary toe excessive amyloid deposition in the extracellular matrix of the myocardium. The gold standard test for diagnosis remains the tissue biopsy, which can be either endomyocardial or fat pad biopsy• Management relies on treating underlying plasma cell dyscrasias with chemotherapy and stringent symptomatic treatment as figure 2 [2].• The median survival may improve to 17 months with immunomodulators and steroids [3].• High-sensitivity cardiac troponin T (hs-cTnT), NT-proBNP, cardiac troponin I, left atrial enlargement, right ventricular dysfunction, and syncope are all poor prognostic factors for AL type cardiac amyloidosis [4].
Conclusions: Cardiac involvement in AL type amyloidosis imparts significant morbidity and mortality to patients suffering from plasma cell dyscrasias. The management of cardiac amyloidosis entails a multidisciplinary approach, particularly cardiology and oncology. Despite the novel diagnostic modalities and treatment regimens, the outcome for AL type cardiac amyloidosis remains poor.