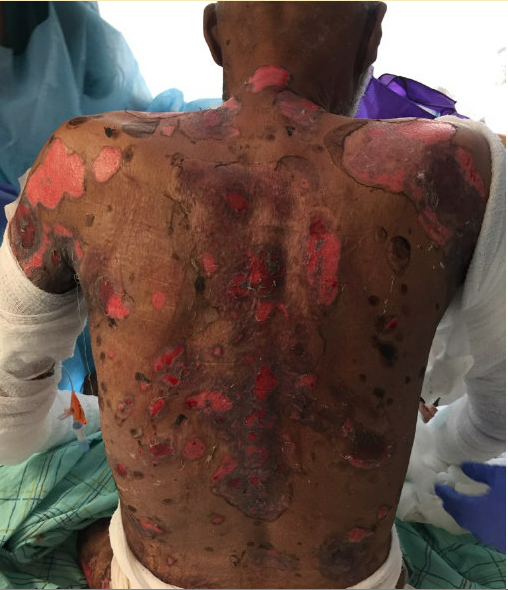
Case Presentation: 63M with severe primary closed angle glaucoma, DMII, pemphigus vulgaris (PV) presents to ED with skin lesions over most body. Functional status impaired due to lower extremity edema, oral lesions resulting in odynophagia and malnutrition. PV recently diagnosed via skin biopsy on H&E stain without immunofluorescence following new blisters beginning on tongue, hard palate, spreading symmetrically to face, extremities, and trunk. Treated with Prednisone, however, worsened glaucoma and diabetes. Attempts to taper resulted in flare; transitioned to Azathioprine without improvement despite escalated doses. Switched to Methotrexate weekly, which halted development of new lesions. However, existing lesions enlarged, denuded, and malodorous, prompting visit to ED.
In ED, affected BSA ~30% with eroded skin on scalp, trunk, axilla, extremities, scrotum, and gluteal creases; oral mucosa; and fluid-filled blisters on right hand, bilateral axilla. Started on broad spectrum antibiotics due to high risk of infection. Dermatology, ophthalmology, and ENT teams consulted with a consensus to avoid corticosteroids in the setting of severe glaucoma. Course complicated by genital HSV swab from groin; completed 10 days of Acyclovir IV with continued prophylactic dosing. Received two doses of Rituximab two weeks apart with weekly MTX; clobetasol, mupirocin ointments; and IVIG for three days. Oral lesions and odynophagia treated with dexamethasone swish and spit. Discharged to subacute rehabilitation for continued wound care. Outpatient course with improvement in lesions. Maintained on weekly MTX and monthly IVIG.
Discussion: Pemphigus is an autoimmune disease characterized by blistering of skin and oral mucosa due to IgG and C3 complements binding to desmoglein adhesion molecules, resulting in acantholysis, epidermal fluid accumulation, and blisters. PV is the most common form with lesions that began orally and spread to face, scalp, and axilla. Onset is most common at 40-50 years of age and has genetic and environmental components. Diagnosis is clinical and histological.
Conventional treatment for PV is immunosuppression via corticosteroids with adjunctive azathioprine and/or mycophenolate mofetil. This case presents challenges associated with corticosteroids: glaucoma and diabetes. Prednisone worsened both conditions, prompting alternative therapy.
In June 2018, the FDA approved Rituximab for PV. This Anti-CD20 monoclonal targets the CD-20 antigen of B lymphocytes, altering cell-cycle differentiation and activation. The International Bullous Disease Consensus Group’s guidelines that indicates corticosteroids and anti-CD20 monoclonal antibodies as first line therapy. Findings based on Ritux 3 trial that included Rituximab with short term systemic steroids or long-term immunosuppressive treatment resulted in remission among 90% of subjects. Adverse events included infusion reaction, depression, and infections: HSV, HZ, UTI, fungal infections, bronchitis, and conjunctivitis. Additional recommendations include corticosteroid-sparing agents including Azathioprine, Mycophenolate mofetil, and IVIG.
Conclusions: The development of biologic agents has permitted targeted therapy and precision medicine, particularly in the treatment of autoimmune diseases. In this case, Rituximab and IVIG acted as steroid sparing therapy in the management of severe PV, decreasing the use of steroids and aggravation of comorbidities.