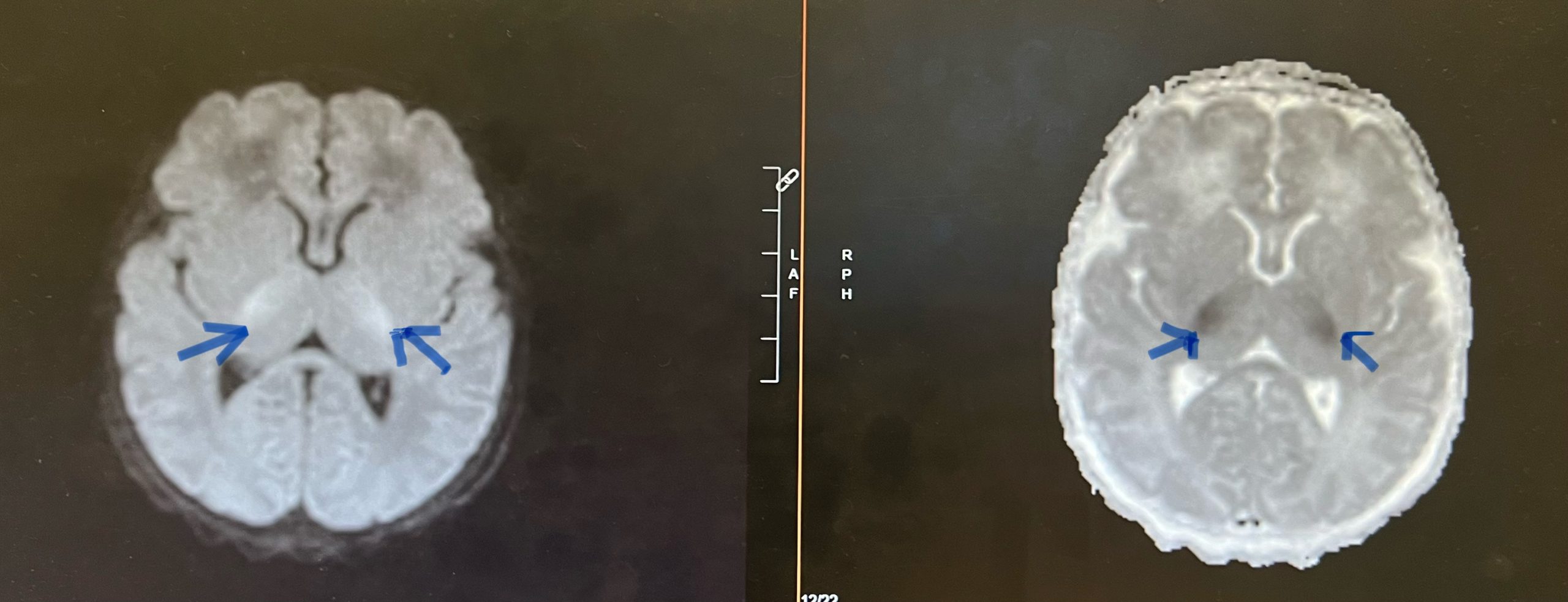
Case Presentation: A two-day old exclusively breastfed male born at term (no prior medical history; routine prenatal care) was discharged at 36 hours of life from the nursery, 5% down from birth weight. He had difficulty latching to feed post discharge and at 45 hours of life, presented to an outside emergency room in cardiorespiratory arrest.Pupils were fixed and dilated, and he was pulseless in the outside emergency room waiting area. He was resuscitated with return of spontaneous circulation. He was placed on an epinephrine infusion. Upon arrival to the Pediatric Intensive Care Unit, initial vital signs while on the epinephrine infusion were as follows: heart rate of 150, respiratory rate of 48, blood pressure of 96/81, and temperature of 98.7 degrees Fahrenheit.Initial glucose was “too low to read” and arterial blood gas only reported pH < 7. Echocardiogram showed severely depressed left ventricular function. Brain MRI showed diffusion restriction in the basal ganglia region suggestive of hypoxic ischemic encephalopathy. Sepsis workup was negative, lactic acid was 5.7 μmol/L, urinalysis with small ketones (10mg/dl), and acylcarnitine profile strongly suggestive of Medium-Chain Acyl-Coenzyme A Dehydrogenase Deficiency (MCADD). State Newborn Screen resulted on day 12 of life, indicative of MCADD-C8:17.7, C10:1.43, C8/C10: 12.37. Hypoglycemia panel genetic testing returned positive. Gene: acyl-Coenzyme A dehydrogenase (ACADM) variant: c.985A>G (p-Lys329Glu), zygosity: homozygous.
Discussion: MCADD is an atypical etiology of cardiorespiratory arrest in the pediatric population. It is a rare autosomal recessive inborn error of metabolism, challenging to diagnose early in life (prevalence: 1/50,000 births) [1]. MCADD is caused by a decreased level of medium chain acyl-coenzyme A dehydrogenase enzyme, which breaks down fatty acids for energy. Individuals with MCADD appear phenotypically normal at birth with onset of presentation between 12 months and 3 years of age. The disorder may be asymptomatic; it is commonly characterized by hypoketotic hypoglycemia, seizures, and lethargy. In most severe cases, undiagnosed MCADD can lead to cardiac arrest and death, so early recognition is key to treatment response and overall prognosis [1].Levocarnitine (L-Carnitine), an amino acid derivative, may be depleted in MCADD. Supplementation helps shuttle fatty acids across the mitochondrial membrane to make energy and prevent metabolic crisis in MCADD [2]. Riboflavin helps make adenosine triphosphate (ATP), needed for fatty acid oxidation [3]. MCADD is hard to identify, especially within the first few days of life, and can mimic neonatal sepsis.
Conclusions: Neonates with MCADD are at risk of metabolic crisis, cardiopulmonary arrest, and death [1]. Newborn screens test for inborn errors of metabolism such as MCADD; however, the results do not return until weeks after birth. This case provides evidence supporting the consideration of MCADD in the differential of a primarily breastfed, term newborn who presents with severe hypoglycemia and cardiorespiratory arrest, particularly with no identifiable risk factors known to cause neonatal hypoglycemia. It also raises an important question of whether hospitals should consider revising or implementing new guidelines/protocols to include closer glucose monitoring for exclusively breastfed infants who are experiencing feeding difficulties in the nursery, especially considering increased rates of exclusive breastfeeding.