
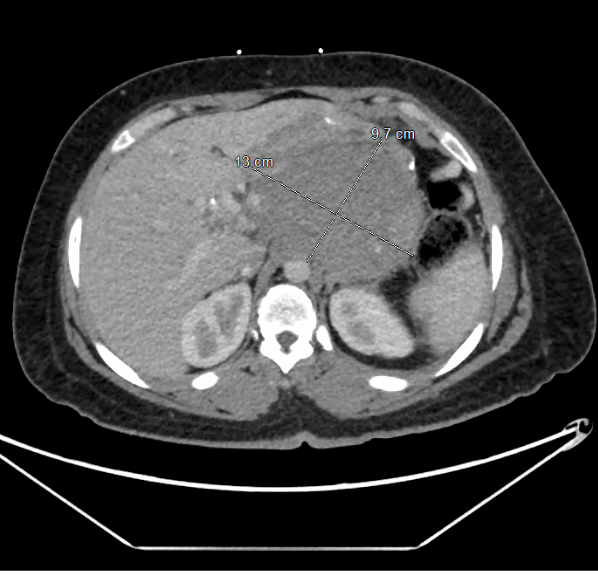
Case Presentation: A 35-year-old female with a past medical history of sleeve gastrectomy presents with abdominal pain, vomiting, and poor oral intake. Objectively, vital signs were stable with mild generalized abdominal tenderness on examination. Laboratory studies including complete blood count and comprehensive metabolic panel were grossly unremarkable. CT abdomen demonstrates a pancreatic mass 9.4 x 10.5 cm enveloping several neighboring vessels. A subsequent diagnostic laparoscopy with biopsy was performed. Histopathology reveals myofibroblastic type proliferation with immunohistochemistry markers consistent with desmoid fibromatosis. A multidisciplinary discussion was held, and the tumor was deemed not suitable to operative excision or radiotherapy. Hence, the patient elected to proceed with systemic therapy consisting of nirogacestat. She was discharged in a hemodynamically stable condition. Nine months later, she presents with worsening abdominal pain, nausea and vomiting. CT abdomen discloses progression of the pancreatic mass to 13 x 10 x 9.7 cm in size. The mass encases the celiac artery and obstructs the splenic vein with narrowing of portal vein. The stomach is severely compressed. She subsequently underwent upper gastrointestinal endoscopic ultrasound disclosing a heterogenous mass in pancreatic body which was sampled through fine needle aspiration biopsy. Histopathological results demonstrated bland spindle cell proliferation compatible with pancreatic desmoid fibromatosis. In view of the patient’s poor nutritional oral intake, she underwent jejunostomy tube placement for enteral nutrition. Systemic therapy was transitioned to liposomal doxorubicin 40 mg/m2 every 4 weeks with good tolerability. The patient was discharged in a stable condition. Three months later, a follow up MRI of the abdomen demonstrates a positive treatment response. She remained on liposomal doxorubicin with close outpatient oncology follow ups.
Discussion: Desmoid tumors represents a group of soft tissue neoplasms that arise from clonal proliferation of myofibroblasts. It comprises 0.03% of all neoplastic processes with an incidence rate of 2-4 cases per million per year. Pancreatic desmoid tumors are exceptionally rare, with only a few published case reports present in the literature. Although they lack a malignant potential, they do have a propensity to invade surrounding structures. The etiology is unclear, however prior associations involved intra-abdominal trauma (e.g. surgery), hormonal, and genetic factors. Our patient underwent a sleeve gastrectomy 2 years prior to diagnosis which could be an inciting factor. Although a multimodal therapeutic regimen is preferred, our patient was not a candidate for surgical excision owing to the extensive nature of her desmoid tumor and the high risk of recurrence in reproductive age. After switching her cytotoxic therapy from nirogacestat to liposomal doxorubicin, a notable treatment response was evident on follow up abdominal imaging.
Conclusions: Pancreatic desmoid tumors are rarely encountered in clinical practice. They do present with compressive symptoms secondary to their local invasion of neighboring tissues. A multimodal management plan including surgical and systemic cytotoxic therapy is always guided by tumor size, extension and patient’s co-morbidities. Further longitudinal studies are needed to establish long-term prognosis of pancreatic desmoid fibromatosis.
.png)