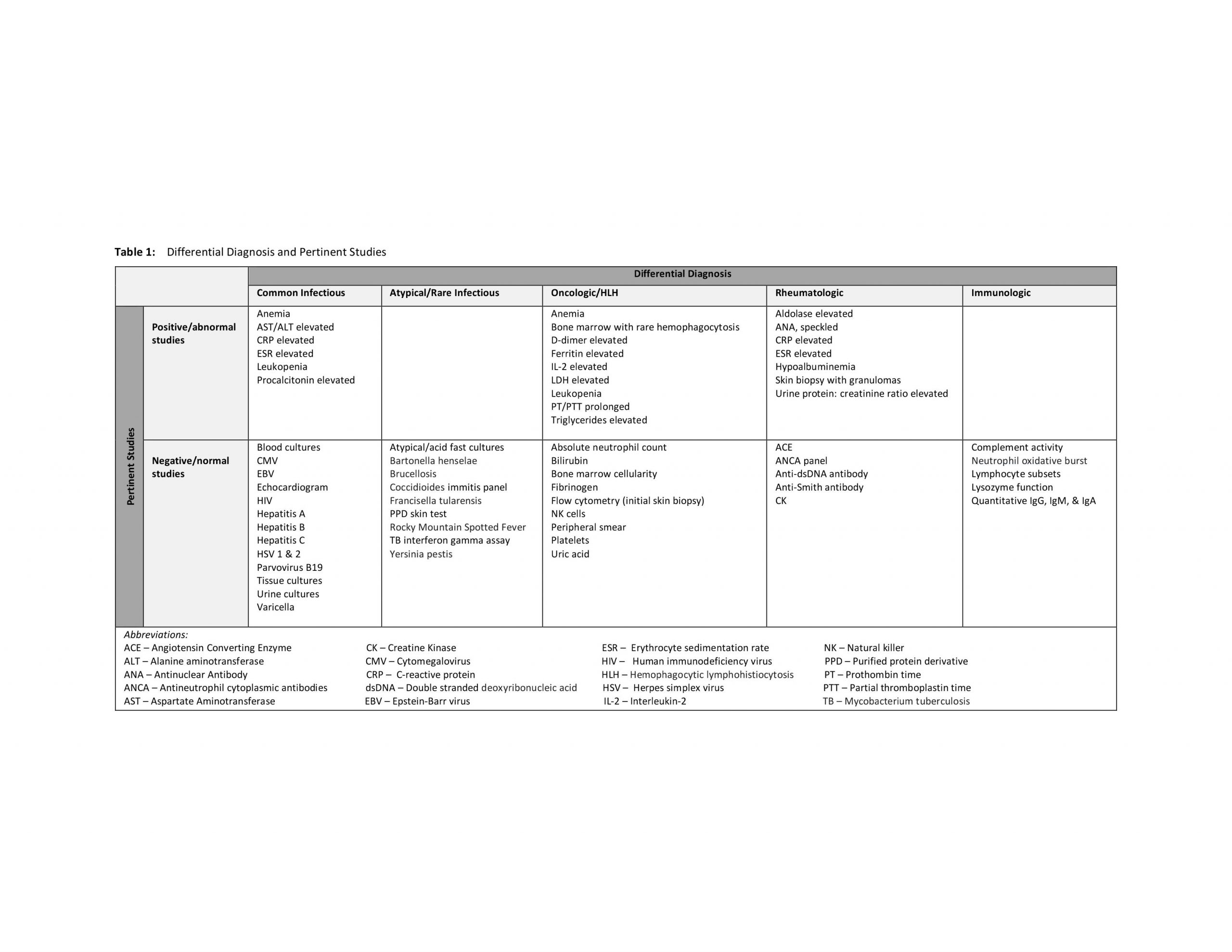
Case Presentation: A previously healthy 10 year-old girl was admitted directly to the pediatric ward with several weeks of fever of unknown origin, lower extremity edema, right groin edema, erythema and pain. She frequently traveled to Mexico and lived in a farm-type setting.She had been treated for cellulitis with at least 7 different antibiotics at outside facilities without resolution of symptoms. Right groin incision and drainage with biopsy performed at an outside hospital demonstrated inflammatory and vascular changes, fat necrosis, scattered epithelioid and giant cell granulomas. Flow cytometry on the biopsy specimen was negative for Non-Hodgkin’s B or T cell lymphoma. On presentation she was febrile and in compensated shock. Exam revealed a painful, indurated, right inguinal cellulitis and tense bilateral lower extremity edema. No lymphadenopathy or organomegaly was appreciated and the skin exam was otherwise negative. Differential diagnosis was broad and extensive workup was initiated with eventual involvement of many subspecialists (table 1). Antibiotics were continued initially, without alleviating symptoms. Bone marrow biopsy revealed rare histiocytes performing erythrophagocytosis, yet labs never fully met criteria for HLH. Hyperalgesia, tachycardia and daily fevers (Tmax 42° C) were persistent despite interventions and she briefly required vasopressor support. She eventually developed a red/violaceous maculopapular rash that began in the axilla and spread diffusely. She underwent a repeat skin biopsy of the new rash and examination was consistent with Subcutaneous Panniculitis-like T-Cell Lymphoma. She was transferred to the Oncology service and treated with prednisone and etoposide, which lead to resolution of her fevers. Outcome is unavailable as care was transitioned to an oncologist in Mexico.
Discussion: Subcutaneous Panniculitis-like T-Cell Lymphoma (SPTCL) is a rare, cytotoxic T-cell lymphoma of the skin layers, often associated with hemophagocytic syndrome, and is estimated to comprise <1% of all Non-Hodgkin’s lymphomas. SPTCL presents most commonly in young adults (median age at diagnosis is 39 years), occurs twice as often in females, and is especially rare in children. Patients may present with either an indolent or aggressive clinical course including prolonged fevers and deep-seated skin nodules or plaques.Diagnosis is challenging, especially in early stages as the symptoms range from mild to severe and may mimic other common and/or benign conditions such as eczema or lipomas. Initial biopsy findings may be misinterpreted as benign.Given its rarity in childhood, there are no specific recommendations for SPCTL therapy and treatments run the spectrum of monotherapy to multi-drug chemotherapy followed by autologous stem cell transplantation. Outcomes for SPTCL vary depending on immunophenotype, α/β vs. ϒ/δ, with estimated 5-year survival rates of 82% and 11%, respectively.
Conclusions: Subcutaneous Panniculitis-like T-Cell Lymphoma is a rare cutaneous lymphoma estimated to comprise <1% of all Non-Hodgkin’s lymphomas. The 5-year overall survival rates vary and are predominantly dependent on immunophenotype. While extremely rare in children, SPTCL should remain on the differential diagnosis of fever of unknown origin for the pediatric hospitalist. Given that biopsy with immunohistochemical analysis is required to definitively diagnose, early suspicion and thoughtful involvement of consultants are key for timely diagnosis and treatment.