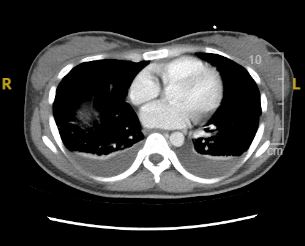
Case Presentation: A 26-year-old female with a past medical history of idiopathic thrombocytopenic purpura (ITP) presented to the hospital with a two-month history of progressive abdominal distension and nausea. On physical examination, she had decreased breath sounds at the bilateral lung bases and massive ascites. Laboratory tests revealed normocytic anemia, mild hyponatremia, slightly elevated creatinine level, hypoalbuminemia, elevated ESR, CRP, and CA-125 (83 U/mL). Beta HCG, liver function tests (LFTs), and hepatitis panel were unremarkable. The gastroenterologist ordered a diagnostic and therapeutic paracentesis, which drained 2 liters of clear fluid, with sterile culture, benign cytology, and a serum ascites albumin gradient of < 1.1. Echocardiogram showed normal ejection fraction. Considering elevated CA-125 with ascites, detailed gynecological examination and diagnostic imaging including abdominal ultrasound, CT abdomen and pelvis, MRI, and endoscopy were done which revealed moderate ascites, mild bilateral pleural effusions, and mild hepatomegaly, with no pelvic masses. Urinalysis showed 3+ protein and 1+ blood, prompting further immunological evaluation for nephrotic syndrome. Immunological workup, guided by nephrology and rheumatology consultations, revealed positive Anti-Smith antibodies (>8), Anti-ds DNA antibodies (265 IU/mL), Anti-SSA, and Anti-RNP antibodies, along with low C3 and C4 complement levels. The patient tested negative for ANA, ANCA, Anti-CCP, and Anti-phospholipid antibodies. A renal biopsy confirmed the diagnosis of class V lupus nephritis. The patient was started on intravenous steroids and subsequently discharged with a regimen including steroids, mycophenolate mofetil, and hydroxychloroquine after significant clinical improvement.
Discussion: Pseudo-pseudo Meigs syndrome (PPMS) or Tjalma Syndrome is a rare manifestation of systemic lupus erythematosus (SLE). It is characterized by a triad of pleural effusions, ascites, and elevated CA-125 levels in the absence of ovarian tumors which distinguishes it from the classic Meigs syndrome. Although serositis is one of the diagnostic criteria for SLE, the presence of progressive massive ascites with elevated CA-125 as the initial manifestation can be misleading, raising strong clinical concern for gynecological malignancy. It can result in unnecessary diagnostic testing such as imaging, biopsies, or even exploratory surgeries. In our case, the patient’s presentation initially suggested a gynecological etiology. However, further workup, including imaging and serological tests, revealed no evidence of ovarian tumors but rather pointed toward SLE as the underlying cause. Furthermore, no special management of PPMS is required other than management of SLE itself and the prognosis largely correlates with the disease activity of SLE. Therefore, early recognition of SLE is crucial, as prompt initiation of immunosuppressive therapy can lead to stabilization and effective management of SLE-related complications.
Conclusions: This case highlights the importance of maintaining a high clinical suspicion for systemic lupus erythematosus, especially in the presence of atypical manifestations such as Pseudo-Pseudo Meigs Syndrome. Although it mimics Meigs Syndrome, the correct diagnosis is crucial to avoid unnecessary invasive investigations for benign and malignant pelvic etiologies and delay the management of SLE, leading to complications.