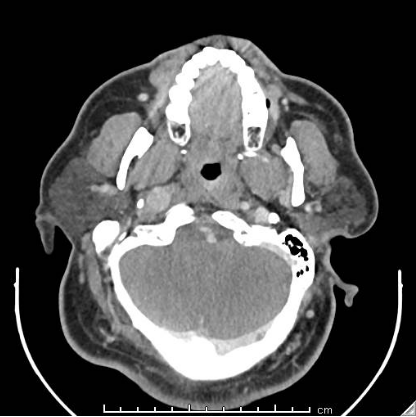
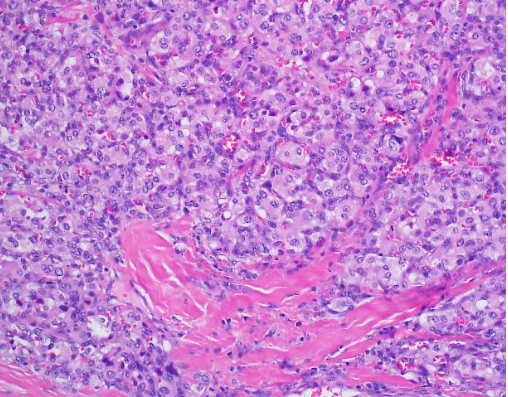
Case Presentation: We report the case of a 43-year-old man who presents to our institution with recurrent episodes of dizziness, headaches and syncope. He had a history of uncontrolled hypertension that was diagnosed as a teenager. His admitting blood pressure was 174/103 mmHg and he had a heart rate of 108. On physical exam, he was obese and appeared uncomfortable. He had some mild, bilateral neck swelling. Biochemical evaluation resulted in urine and plasma metanephrines. A CT scan of the neck’s soft tissue revealed a 0.8 cm right carotid body tumor, a 1.7 cm left carotid body tumor, and a 1.2 cm right glomus vagale tumor, suspicious for paragangliomas (PGLs). Given the magnitude of this patient’s symptoms and lab findings, surgery was performed. Initially, the right carotid body tumor was embolized by Neurosurgery and then Otolaryngology did a right neck dissection to remove the right carotid body and glomus vagale tumors. While the patient did have improvement in his symptoms, he unfortunately developed right facial nerve paresis, a common surgical complication. Histopathologic analysis returned consistent with PGLs and negative for malignancy. Interestingly, the patient’s brother also had a similar presentation in his 40’s consisting of headaches and dizziness. He was diagnosed with isolated bilateral carotid PGLs and subsequently underwent surgery and radiation. Unfortunately, his brother was at another institution and due to insurance issues, was unable to receive genetic testing. Considering the family history of PGLs, mutations for our patient were sent for succinate dehydrogenase (AF2, A, B, C and D) as well as Neurofibromatosis-1 (NF-1). He tested heterozygous for the presence of a Succinate Dehydrogenase complex subunit D (SDHD) c.13_14delTG pathogenic mutation, consistent with the diagnosis of Hereditary Paraganglioma-Pheochromocytoma Syndrome type 1. This pathogenic mutation consists of a frameshift resulting in a truncated protein.
Discussion: PGLs are rare, typically benign tumors that originate from neuroendocrine tissue. They have an incidence of 1 in 300,000 per year. Up to 35% of these tumors can be hereditary and the majority are associated with germline mutations in genes encoding subunits of the succinate dehydrogenase enzyme complex. Less frequently, mutations in the genes responsible for Von Hippel Lindau disease, Multiple Endocrine Neoplasia type 2, and Neurofibromatosis type 1 are also found in patients with hereditary PGL and Pheochromocytoma. The carotid body is the most common location for PGLs and they are frequently multifocal in origin. While there are no specific guidelines for treatment of bilateral head and neck PGLs, therapeutic options include surgical resection, radiation therapy, stereotactic radiosurgery, embolization, or a combination of these modalities.
Conclusions: The patient we described in this case developed worsening postural dizziness and headaches over the course of 20 years. Multiple resources were utilized before arriving at the diagnosis of bilateral carotid body PGLs. Prior literature states that PGLs of the head and neck do not typically produce catecholamines and are therefore diagnosed late when a mass effect syndrome occurs on nearby organs. While the majority of head and neck PGLs are non-secreting, our case emphasizes the possibility that secretory ones may be more common than previously thought. Further research is needed to gain a better understanding of bilateral head and neck PGLs.