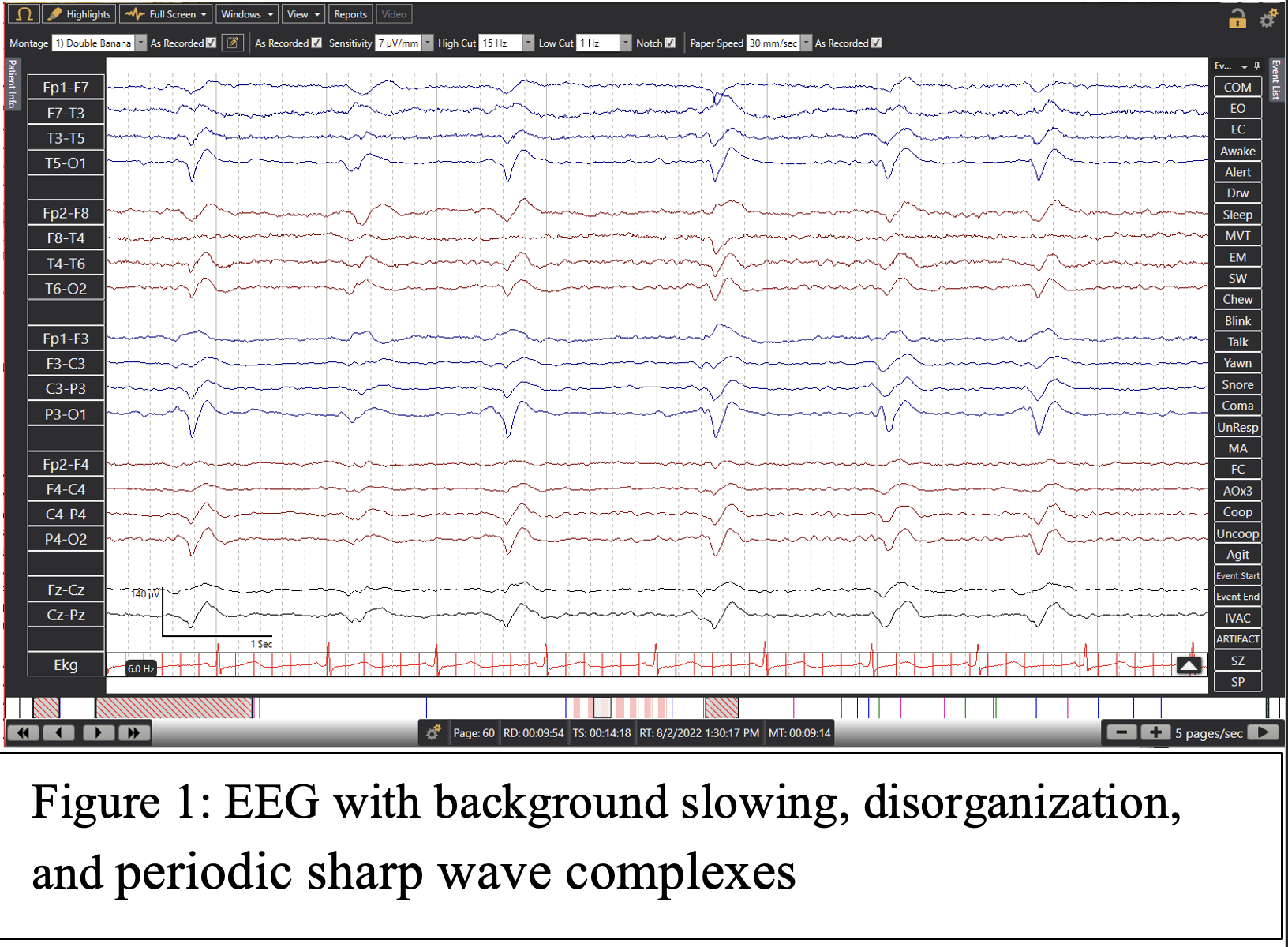
Case Presentation: A fifty-three year old male presented with alcohol intoxication, generalized fatigue of two days, hyperglycemia and lack of appetite. He was ill-appearing, tachycardiac, severely dehydrated and distended. The patient had a long history of chronic ethanol abuse, liver disease, recurrent anemia, a right frontotemporal hemorrhage, and was found to have pancreatitis and encephalopathy on this admission. Throughout the hospitalization he had periods of memory loss, incomprehensible speech, incontinence, confabulations, and seizure-like activity for which the patient was started on Keppra. (6)An electroencephalogram (EEG) performed indicated a degree of bihemispheric dysfunction. (Figure 1) Magnetic resonance imaging (MRI) of the brain revealed a stable right temporal bleed and cortical ribbon sign in the left parietal region (Figure 2a-2b) with increased FLAIR signal in the basal ganglia and thalamus (Figures 2c-2e).Lumbar puncture was done to check for protein 14-3-3 and to perform real-time quaking-induced conversion assay (RT-QulC) to determine CJD versus non-CJD origin. (5) Though the latter results were indeterminate, 14-3-3 was seen elevated in the CSF supporting the diagnosis of CJD. The patient’s dose of Keppra was then adjusted to a higher dose and hospice palliative care was initiated for the comfort of the patient.
Discussion: Prion proteins (PrP) exist in the cellular form PrPc or the pathological form PrPsc. While PrPc has an alpha helical secondary structure similar to the native protein, PrPsc has a beta pleated secondary structure in specific regions. This structure makes it more likely to aggregate and converts the structure of PrPc to beta pleated causing spongiform degeneration. (7)PrPsc in acquired CJD originates through infection while sporadic CJD is theorized to initiate through a mutation that develops as a result of the aging process or spontaneous conversion. Familial CJD presents with a pattern of inheritance of mutated PRNP gene that encodes for prion proteins and can cause an extensive array of diseases depending on the mutation. (8)Magnetic resonance imaging (MRI) is the gold standard of imaging for diagnosis and included fluid attenuated inversion recovery (FLAIR) sequence with diffusion weighted imaging (DWI) helps determine the acuteness of the cerebral event being evaluated. (2) The presence of cortical ribboning, hyperintense signal changes in the gray matter, is also a finding supportive of CJD though not specific to prion diseases. (3)CSF evaluation for protein biomarkers 14-3-3, total tau (t-tau) and neuron-specific enolase (NSE) are all supportive of neurodegeneration. Real Time-Quaking-Induced Conversion (RT-QulC) is very specific for CJD and highly sensitive in the detection of prion proteins in the CSF. (4)Electroencephalogram (EEG) with periodic sharp wave complexes and a degree of disorganization is supportive of CJD. A confirmatory diagnostic study for CJD would be a brain biopsy, however due to the invasive nature of this test and its lack of necessity after other supportive diagnostic findings, it is not always performed. (2)
Conclusions: Due to the few cases and even less literature available on the disease, CJD may often be missed as a potential differential for a patient presenting with symptoms of dementia and debilitating neurologic symptoms. Although prognosis is poor and there is no current treatment, assuring the comfort and palliative care of a patient early in their clinical course is greatly significant and a worthwhile goal.