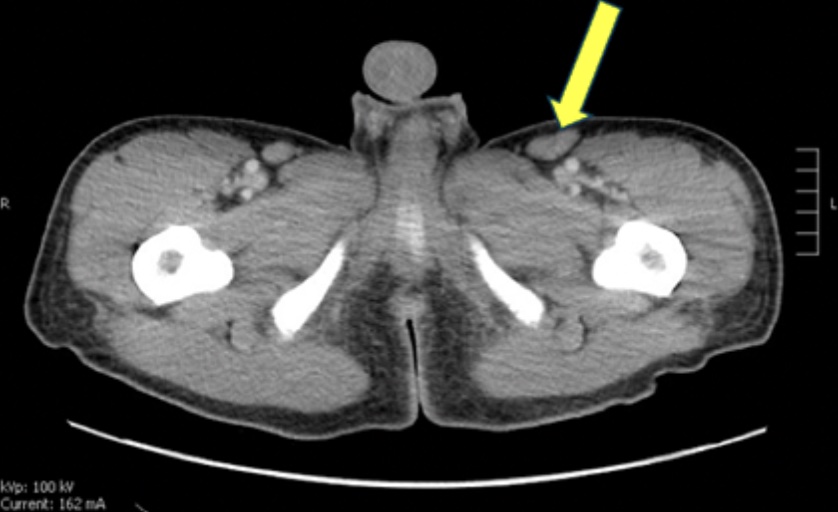
Case Presentation: A 33-year-old healthy male presented to Yale New Haven Hospital with a 3-week history of fevers up to 103°F, drenching night sweats, rigors, fatigue, mental fog, and 20lb weight loss accompanied by poor appetite. He denied recent travel or sick contacts. Recent outside hospital evaluation included negative blood cultures, Lyme Ab, EBV, hepatitis A/B/C, and anaplasma testing. Lumbar puncture showed low glucose and mild protein elevation with negative CSF culture. Chest CT revealed apical pleural-parenchymal scarring and diffuse mediastinal and axillary lymphadenopathy. The patient was prescribed a 7-day course of doxycycline and amoxicillin-clavulanate, with no improvement of symptoms. On return admission, he was febrile to 102.6°F and tachycardic, with diffuse tender lymphadenopathy and nonpruritic rash including the palms and soles. CT demonstrated mediastinal, hilar, supraclavicular, axillary, retroperitoneal lymphadenopathy, splenomegaly and pulmonary ground glass opacities. Lab findings included hyponatremia of 129, elevated transaminases (AST 77, ALT 62), CRP 40.2, and LDH 617. Infectious disease, rheumatology, and hematology were consulted. Work-up included HIV, EBV, CMV, Bartonella, fungal antigens, ANA subtypes, flow cytometry, SSA, SSB, Smith, RNP, DsDNA, C3, C4, immunoglobulins, and tularemia testing, which was unrevealing except for a positive ANA (≥1:2560). General Surgery performed an excisional left inguinal lymph node (LN) biopsy. Patient’s fevers were controlled with acetaminophen and rash improved spontaneously. After discharge, LN biopsy returned with paracortical hyperplasia with Castleman-like changes. Outpatient axillary excisional LN biopsy performed to ensure no lymphoma present showed reactive lymphadenopathy without malignancy. Serum interleukin-6 (IL-6) was elevated and HHV-8 serology was negative, consistent with HHV-8 negative Multicentric Castleman Disease. His fevers and constitutional symptoms gradually resolved without targeted therapy following the additional LN removal.
Discussion: Castleman Disease is a rare, nonclonal lymphoproliferative disorder characterized by angiofollicular LN hyperplasia and IL-6 hyperexpression. Unicentric disease, confined to a single LN region, is typically curative with surgical excision. In contrast, multicentric disease (MCD) involves multiple LN stations and manifests with systemic inflammation, hepatosplenomegaly, cytopenias, and organ dysfunction. Autoimmune markers such as positive ANA may be seen, particularly in idiopathic MCD. Management of MCD depends on HHV-8 status. HHV-8 negative MCD is treated with IL-6 targeted agents: siltuximab or tocilizumab. Patients with HHV-8 associated MCD and/or compressive lymphadenopathy can receive rituximab. Uniquely, despite diffuse clinical and radiographic LN involvement, this patient’s MCD symptoms improved following second LN excision without a pharmacologic agent.
Conclusions: This case illustrates the diagnostic and therapeutic complexity of Castleman Disease. MCD can present to the inpatient setting with nonspecific systemic inflammatory features that mimic infection, malignancy, and autoimmune disease, underscoring the need for awareness among hospitalists. The atypical improvement of MCD with LN excision alone observed with this patient raises important questions about the underlying mechanisms driving disease remission and highlights the need for further research.