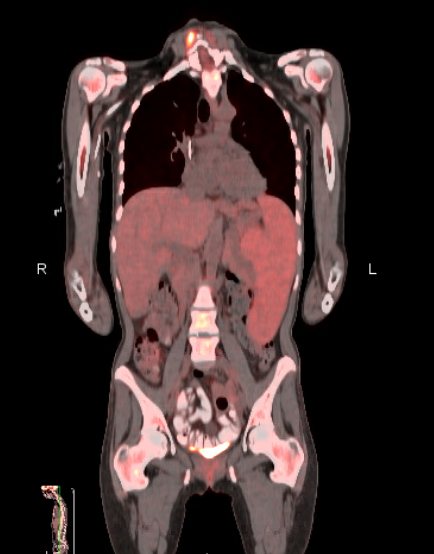
Case Presentation: A healthy 38 yo woman presented to hospital with 3 weeks of fatigue. She was recently treated for a urinary tract infection but continued to feel unwell. She complained of mild LUQ abdominal pain. On review of systems she endorsed weight loss, fever and night sweats but denied chest pain, dyspnea, easy bleeding, recent travel or sick contacts.On examination temperature was 101F, heart rate 105 bpm, with a normal blood pressure. In general she appeared well, had clear lung fields and normal cardiac findings. Pronounced splenomegaly and mild hepatomegaly were present. Skin was without rashes.Laboratory findings revealed WBC count of 1.7 x 10^9/L, hemoglobin 6.0 gm/dL, and platelets of 94 x 10^9/L. Total bilirubin was 1.9 gm/dL and LDH of 453 with undetectable haptoglobin. Ferritin was 4053, and soluble IL2 receptor was 9889. CT imaging of chest, abdomen and pelvis revealed splenomegaly and mild hepatomegaly.She remained febrile, pancytopenic, and underwent bone marrow biopsy which was without pathology. PET CT revealed mild FDG uptake in the spleen. Although hemophagocytic lymphohistiocytosis (HLH) was strongly suspected, an underlying cause could not be identified and the absence of pathologic confirmation of hemophagocytosis made the Hematology team reluctant to start chemotherapeutic agents. A broad infectious and rheumatologic work up was negative. After 21 days in the hospital the patient was discharged with follow up but without a diagnosis.She improved but developed pancytopenia and fever within 8 weeks, Repeat PET CT showed an increase in splenic uptake and a splenic biopsy was performed revealing hepatosplenic T-cell lymphoma. Repeat bone marrow biopsy revealed a small clonal T cell population consistent with the same diagnosis.The patient was diagnosed with HLH due to hepatosplenic T-cell lymphoma and was initiated on chemotherapy with plan for allogeneic bone marrow transplantation after remission.
Discussion: Hemophagocytic lymphohistiocytosis is a condition characterized by fever, cytopenias, transaminitis, and hepatosplenomegaly. Although poorly understood it is thought to be a cytokine storm of inflammation induced by hematologic malignancy, autoimmune condition or infection in adults. Diagnostic criteria include five of the following: fever, splenomegaly, cytopenias (2 of 3), hypertriglyceridemia/hypofibrinogenemia, ferritin > 500, low or absent NK cell activity, soluble IL2 receptor >2400 and presence of hemophagocytosis in bone marrow, splenic, hepatic or lymphoid tissue. These criteria were developed for pediatric patients and are of uncertain validity in adults. Hemophagocytosis is seen in 25-100% of cases making it an unreliable finding. Treatment includes dexamethasone and etoposide although again is based on pediatric protocols. Our patient met criteria for HLH but without a clear trigger and absence of hemophagocytosis, treatment was initially withheld. Hepatosplenic T cell lymphoma is a rare hematologic malignancy with infiltration of spleen and liver and is notoriously difficult to identify unless reviewed by an experienced hematopathologist.Prognosis and response to treatment are variable in HLH and the outcomes are better when the triggers are non-malignant. Hepatosplenic T cell lymphoma carries a 5 year survival rate of only 7%.
Conclusions: The hospitalist must remain aware of the diagnostic criteria for hemophagocytic lymphohistiocytosis and keep a high index of suspicion for this difficult-to-define but life-threatening condition.