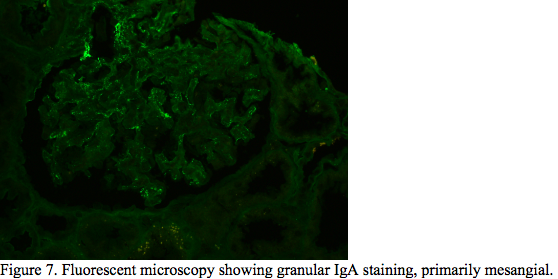
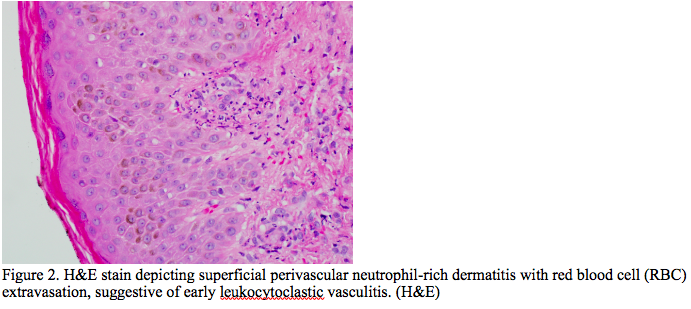
Case Presentation: A 39-year-old male with no known history presented with complaints of back pain and shortness of breath for four days. Work up initially revealed pyelonephritis. The patient was treated with Amoxicillin-Clavunate. After a month, patient was re-evaluated for increased shortness of breath, fatigue, abdominal pain, diffuse arthralgia, and a new rash. The shortness of breath was associated with chest discomfort which worsened upon inspiration and cough with production of white-frothy sputum. Patient endorsed nausea and vomiting, with diffuse abdominal pain, night sweats, headaches, and positional dizziness. Physical examination was positive for abdominal distention and right upper quadrant tenderness on palpation with negative Murphey’s sign. Patient exhibited plus two peripheral pitting edema on bilateral lower extremities. Skin examination was significant for non-blanching, palpable purpuric rash bilateral lower and upper extremities. Lab work up detected a creatinine of 2.36 mg/dL, blood urea nitrogen of 44 mg/dL, alkaline phosphatase of 657 U/L, platelet of 112 10*3 /uL, total bilirubin of 1.7 mg/dL, albumin of 2.5 g/dL. CT scanning showed bilateral pleural effusion, moderate sized pericardial effusion, ascites, hepatosplenomegaly and retroperitoneal lymphadenopathy. ESR and CRP were elevated at 125 mm/hr and 318.2 mg/L, respectively. Autoimmune panel was remarkable for ds-DNA Ab of 153 [IU]/mL, histone antibody of 354 [AU]/mL, anti Jo-1 antibodies of 151 [AU]/mL, anti SSB-Ab of 268 [AU]/mL, and IgG subclass 1 of 917 mg/dL. Tests for infectious etiologies were negative. A skin biopsy was obtained which showed focal fine granular deposition of IgA at small blood vessels of papillary dermis. The findings depicted on the biopsy studies were suggestive of an IgA immunoreactant associated vascular injury/vasculitis. A renal biopsy was obtained which revealed mesangial, subendothelial and intraluminal “pseudo-thrombi” glomerular IgA immune complex deposition. The skin and renal biopsies along with autoimmune panel were consistent with IgA vasculitis with nephritis and SLE.
Discussion: IgA associated vasculitis, most commonly known as a childhood disease, is a small vessel vasculitis associated with IgA immune complex deposition. Its clinical manifestations include palpable purpura, joint, gastrointestinal, and/or kidney involvement. The clinical diagnosis of IgA associated vasculitis relies heavily on histological diagnosis and is supported by clinical signs and symptoms. It is essential for IgA associated vasculitis with renal involvement, also known as IgA vasculitis nephropathy, to be recognized in adults as there is a risk for developing chronic renal failure. Through this case report, the expectation is to not only bring awareness to the possibility of underdiagnosed IgA associated vasculitis and nephritis in adults, but to also explore the possibility of this disease entity carrying a correlation to SLE as an initial clinical manifestation.
Conclusions: SLE is a chronic autoimmune disease that can affect all organs, including the kidneys. The foundation of the disease pathophysiology is associated with antibodies attacking multiple organ systems and causing inflammation, which typically manifests as arthralgias, nephritis, serositis, vasculitis. IgA vasculitis is the most common form of systemic vasculitis in children although it is rarely reported in adults. To our knowledge, IgA vasculitis as a manifestation of SLE has rarely been reported in literature.