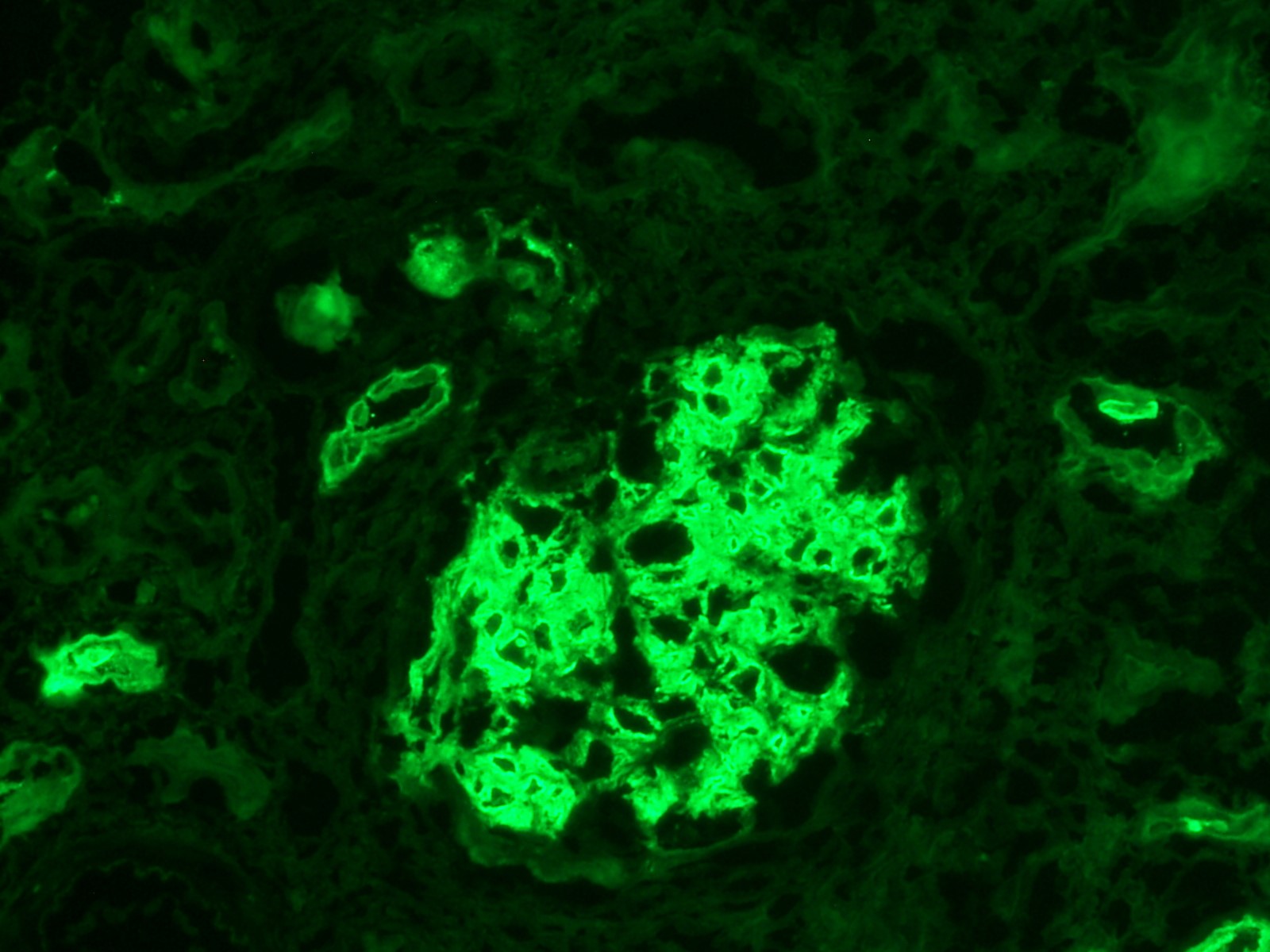
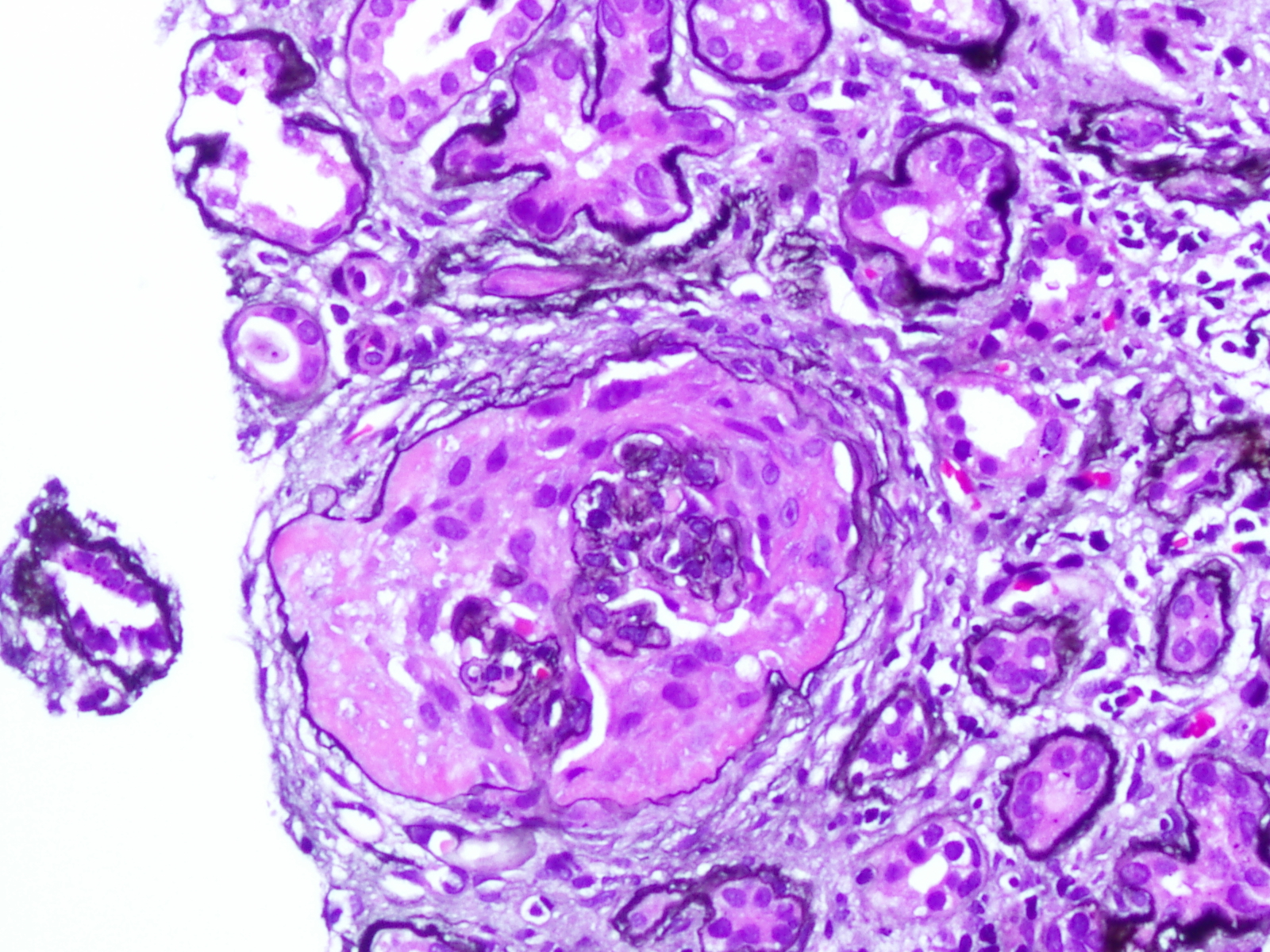
Case Presentation: A 22-year-old male with no past medical history presented with a three-week history of a frontal headache associated with nausea and vomiting. He denied various symptoms, including bloody diarrhea, dysuria, or hematuria. He had no history of renal calculi, recent illness, antibiotic exposure, or drug allergies. He recently traveled to Jamaica and Florida and served in the Army Reserve. Family history revealed his father, who served in Iraq, developed end-stage renal disease necessitating a transplant after a non-diagnostic kidney biopsy. Initial assessment found hypertensive emergency (220/128), elevated creatinine (9.86), BUN (69), and hemoglobin of 9.7. The remaining labs were unremarkable. Urinalysis revealed large occult blood, and >1000 protein. CT head and abdomen/pelvis were negative. He received IV fluids and a nicardipine drip. Nephrology consultation led to a serologic evaluation, including ANA, ANCA, anti-GBM antibody, anti-DS DNA, C3, C4, IgA titers, ASO titers, HIV, CMV, EBV, and E. coli O157. Results yielded negative findings. Serum IgA was 238. Peripheral blood smear and urine protein electrophoresis were unremarkable. Blood cultures showed no growth. Renal US ruled out stenosis. Kidney biopsy revealed IgA nephropathy with endocapillary proliferation, crescentic glomerulonephritis, and evidence of malignant hypertension. Moderate activity, severe chronicity, global sclerosis (20/36) with 70-80% fibrosis, and 7 cellular crescents. Oxford classification: M1, E1, S1, T1-C1. Given the patient’s age and biopsy findings, nephrology favored renal preservation strategies. Treatment included IV solumedrol before prolonged prednisone taper, three rounds of plasma exchange apheresis for crescentic glomerulonephritis, short-term azathioprine to reduce HLA sensitization risk, and short-term hemodialysis.
Discussion: IgA nephropathy arises from the deposition of IgA in the mesangium, leading to hematuria and proteinuria. It commonly manifests initially in young adulthood and is the most prevalent primary glomerulonephritis. Clinical symptoms typically start with benign intermittent hematuria and may not progress any further. For a significant number of patients, IgA nephropathy has minimal impact on their lives. Up to 30% achieve remission, with many maintaining full renal function. However, for 40% of patients older than 10-25 years, glomerulonephritis progresses to end-stage kidney disease. Our patient’s biopsy revealed a severe presentation of IgA nephropathy, demonstrating positivity for each element of the Oxford classification/MEST-C score: mesangial and endocapillary hypercellularity, (global) segmental glomerulosclerosis, interstitial fibrosis/tubular atrophy and crescents. According to KDIGO treatment guidelines, management should encompass supportive care, RAS blockade, BP control, lifestyle and risk factor mediation. If proteinuria persists above 750-1000 mg after 90 days of appropriate care, KDIGO recommends a 6-month course of glucocorticoid therapy. Oral methylprednisolone in IgA nephropathy has shown efficacy in reducing the risk of declining kidney function, kidney failure, and mortality attributable to kidney disease.
Conclusions: This case is a demonstration of severe IgA nephropathy. It illustrates the variability in IgA nephropathy and the need for an individualized strategy to mitigate the risk of progression to end-stage kidney disease.