Case Presentation: A 39-year-old woman with history of migraines, Chiari malformation, and PTSD was admitted with 2 months of constant generalized pain with spasms in her face, neck, shoulders, and back. She had severe and diffuse muscle twitching in her extremities and 6-pound weight loss.At onset of symptoms, MRI of brain and cervical spine showed mild cerebellar tonsillar ectopia. Labs were as follows: normal c-reactive protein, erythrocyte sedimentation rate, aldolase, and Vitamin B12, negative polymyositis and dermatomyositis panel, and creatinine kinase (CK) elevated to 440 U/L. Voltage-gated potassium channel (VGKCs) antibodies were sent. Gabapentin was given with minimal relief.Upon admission, there was concern for fibromyalgia or central hypersensitization given heightened pain complaints and use of high amounts of analgesics. Rheumatology and Neurology were consulted. The patient had diffuse myokymia (involuntary muscular contractions with worm-like appearance), symmetric hyperreflexia, and diaphoresis on exam. Electromyogram (EMG) showed myokymic discharges and grouped repetitive discharges +/- fasciculations. VGKCs antibodies resulted positive. She was diagnosed with Isaac’s syndrome. Computed tomography of chest, abdomen, and pelvis and full body positron emission tomography were unrevealing for malignancy. Lumbar puncture was negative for infectious and inflammatory conditions. She received 5 days of IV Solumedrol followed by a steroid taper. She was seen by Psychology and Psychiatry for history of trauma which exacerbated her pain. The patient improved and was discharged home. She was referred to Pain Management, Neurology, and Neuromuscular Clinic outpatient.
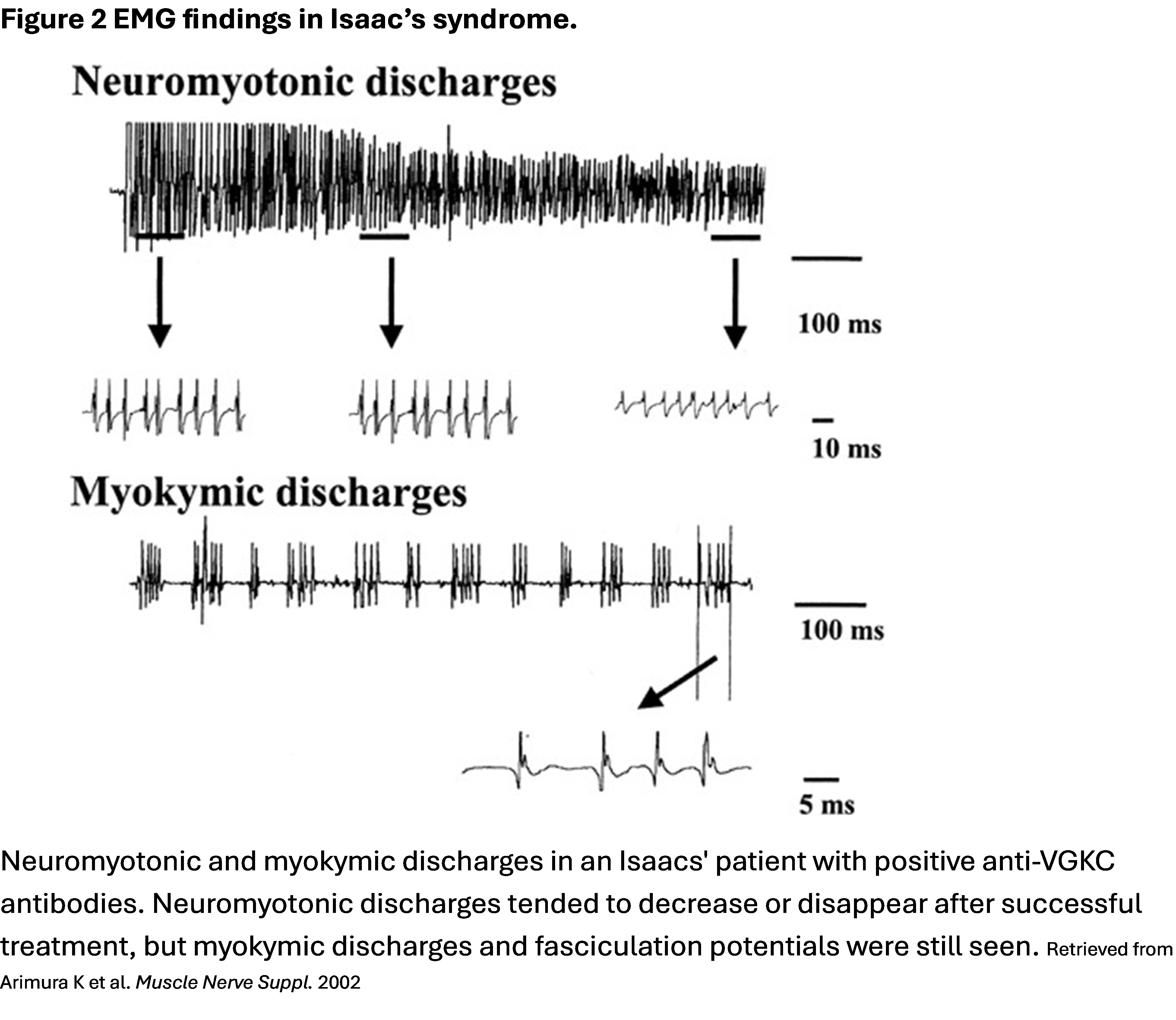
Discussion: Neuromyotonia is a disorder characterized by muscle cramps, muscle twitching including fasciculations and myokymia, and delayed muscle relaxation due to abnormal, spontaneous, and continuous peripheral nerve excitability often affecting the distal limbs. The autonomic nervous system may be involved, leading to hyperhidrosis, orthostatic hypotension, or constipation. Neuropathic pain and paresthesia or other sensory symptoms may occur. Muscle hypertrophy is present in the affected muscle groups and sometimes muscle weakness is present. CK may be elevated. Diagnosis involves clinical, laboratory, and EMG findings (Figure 1).Neuromyotonia is associated with malignancies such as thymoma and small cell lung cancer, autoimmune disease, and with toxin exposure including mercury poisoning. However, the condition can be idiopathic, hereditary, or congenital. Isaac’s syndrome is an often paraneoplastic form of neuromytonia first described by Hyam Isaacs in the 1960s. Antibodies against VGKCs affect 40-50% of cases of Isaac’s syndrome and cause excess acetylcholine at the presynaptic membrane. EMG demonstrates persistent and irregular fibrillations, fasciculations, and spontaneous firing of single motor units (Figure 2). Motor discharges occur during sleep, rest, and even general anesthesia. Treatment relies on immunotherapy and consists of intravenous immune globulin, corticosteroids, and/or plasmapheresis. Antiepileptics and sodium channel blockers, including carbamazepine, and phenytoin, diazepam, or gabapentin may also be used for symptomatic relief.
Conclusions: Isaac’s syndrome is a rare condition that may present as chronic pain. In patients with neuromuscular signs and symptoms such as painful muscle spasms and elevated CK, neuromyotonia should be investigated.