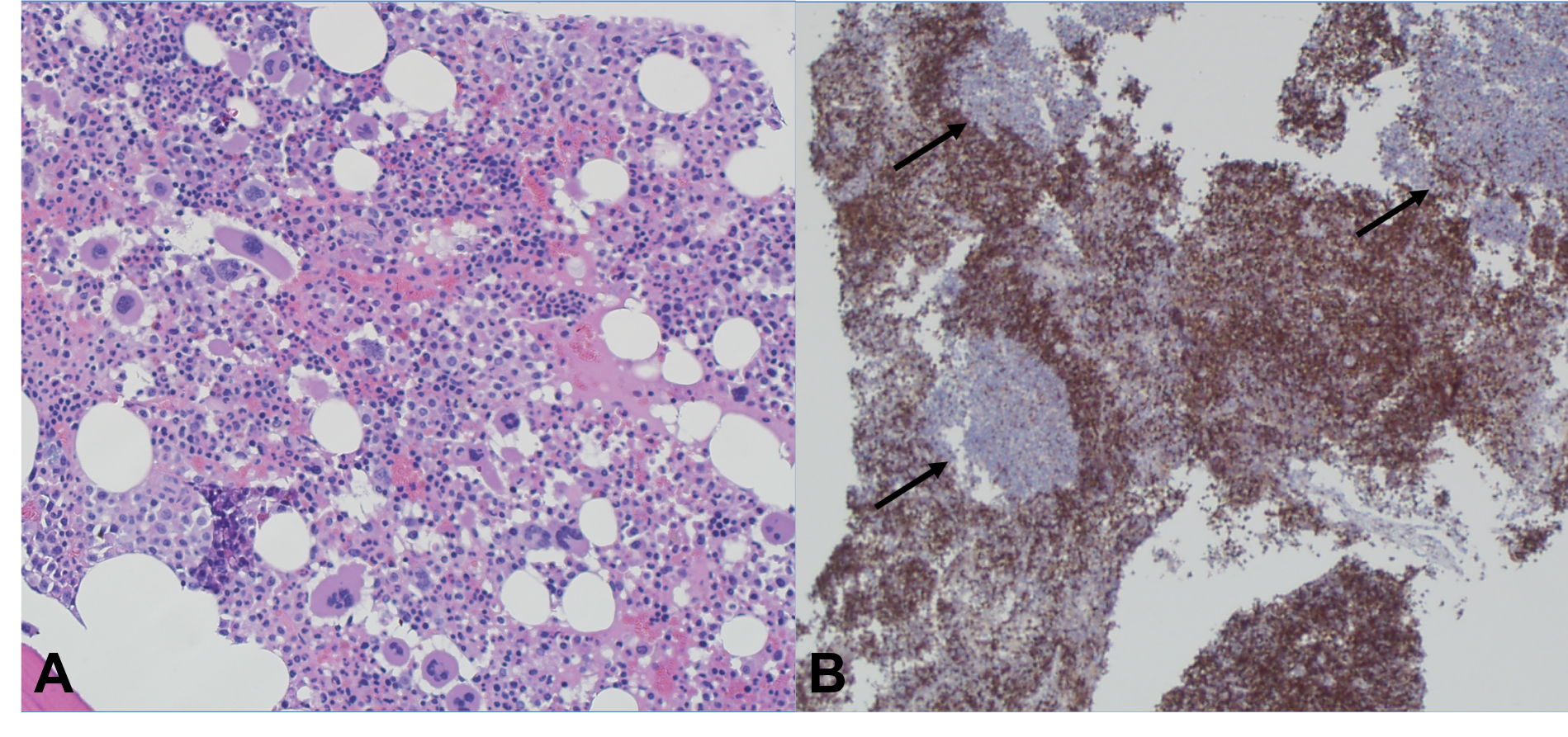
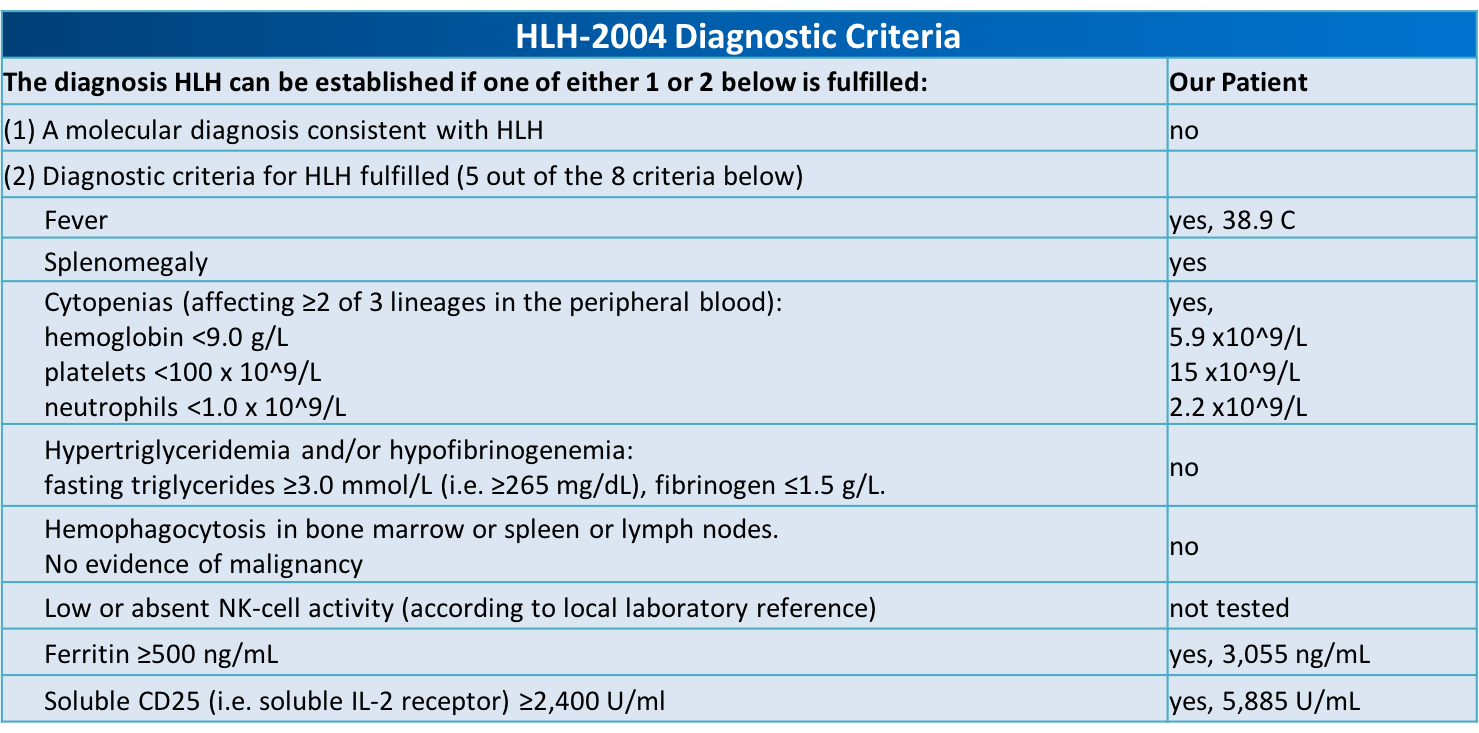
Case Presentation: A 22-year-old male with no known medical history presented with complaints of epistaxis for two weeks and subjective fevers. The patient also reported fatigue, and unintentional weight loss for three months. He was tachycardic to 111 on presentation, but otherwise afebrile and normotensive. Pertinent physical exam findings included dried blood in the nares, presence of petechiae on the upper chest and extremities, abdominal tenderness and a palpable spleen. Laboratory results revealed leukopenia (3.07 [4-10 x 10^9/L]), thrombocytopenia (15 [150-400 x 10^9/L]), and an elevated protein gap. The peripheral smear showed atypical lymphocytes and large platelets with no premature cells. A computed tomography scan of the abdomen showed splenomegaly, left common iliac, left pelvic sidewall, and left inguinal lymphadenopathy.Lymph node and bone marrow biopsies were performed; histopathological analysis was unremarkable. Testing for HIV, hepatitis B, Hepatitis C, cytomegalovirus, parvovirus, and histoplasmosis were negative. Antinuclear antibodies (ANA) and double-stranded DNA (dsDNA) results were unremarkable. Epstein-Barr virus (EBV) serology was significant for prior exposure, however, a positive PCR viral load was oddly noted. A high suspicion of secondary immune thrombocytopenic purpura (ITP) led to treatment with corticosteroids and intravenous immunoglobulin (IVIG). There was a partial response to therapy proven by elevation in platelet count, however, the patient continued to have fevers up to 38.9C and soon developed anemia (hemoglobin 5.9 [13-17 g/dL]) requiring red blood cell transfusions.Confounded at this point, a soluble CD25 test was pursued and found to be elevated (5,885 pg/ml [532-1891 pg/ml]). Given this new finding, along with an elevated serum ferritin (3054.7 ng/mL [30-300 ng/mL]), the patient met 5 of the 8 HLH-2004 diagnostic criteria for Hemophagocytic Lymphohistiocytosis (HLH). He was started on a course of etoposide and dexamethasone with significant improvement and was determined to be in remission after six months.
Discussion: HLH is a rare, life-threatening hyper-inflammatory condition due to overactivation of macrophages and cytotoxic T-cells. The primary form is caused by genetic mutations affecting lymphocyte cytotoxicity and immune regulation and is most commonly seen in infants and children. The secondary form, commonly triggered by infections, malignancies, or autoimmune disorders is more prevalent in adults. Treatment and diagnosis are based primarily on pediatric research and guidelines. Given the rarity of the disease in adults, identification and management may be perplexing. Our patient’s nonspecific presentation and prolonged hospital course makes awareness of this rare hematological disease relevant for hospitalists.
Conclusions: Clinical presentation of HLH in adults can be difficult to distinguish from sepsis, hematological malignancies, or autoimmune disorders. Early diagnosis and treatment are essential to avoid a fatal outcome. EBV is the most commonly associated trigger for secondary HLH. High clinical suspicion should be maintained in patients presenting with fever, bicytopenia, and splenomegaly. Treatment protocols are based on pediatric guidelines, but further investigation is warranted about treatment in adults diagnosed with secondary HLH.