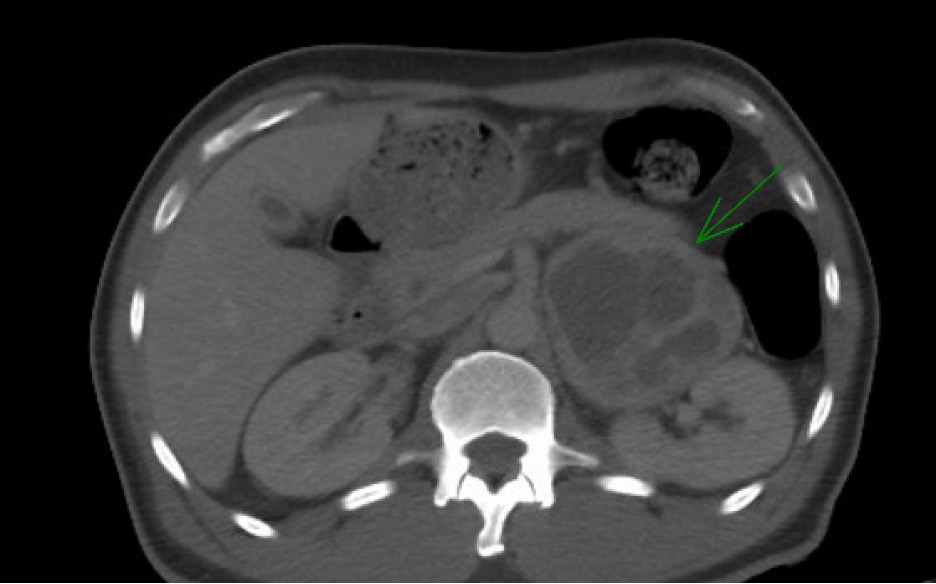
Case Presentation: A 56-year-old man with type 2 diabetes and hypertension presented to the ER with uncontrolled blood sugars, intermittent nausea, vomiting, fevers, and a 15-pound unintentional weight loss over one month. He was recently hospitalized with similar symptoms, started on insulin, and evaluated for fevers with negative blood and urine cultures, which were attributed to a viral illness. At presentation, labs showed severe hyperglycemia, leukocytosis, elevated creatinine, and troponin, but infectious testing including urinalysis and chest X-ray was again unremarkable. A transthoracic echocardiogram revealed diffuse left ventricular hypokinesis with an ejection fraction of 40-44%, prompting initiation of metoprolol. Despite increasing insulin, hyperglycemia persisted. A CT scan of the abdomen and pelvis was performed to evaluate for malignancy, revealing a solid cystic mass in the left adrenal gland measuring 8 x 7 x 9.3 cm. The patient developed hypertensive crises with systolic BP reaching 200 mmHg, alternating with orthostatic hypotension, requiring IV fluids. Suspecting pheochromocytoma, metoprolol was discontinued and phenoxybenzamine was started. Biochemical tests confirmed pheochromocytoma with elevated plasma metanephrine (950 pg/mL), urine metanephrine (7614 mcg/g creatinine), and plasma normetanephrine (875 pg/mL). He underwent left adrenalectomy and nephrectomy due to left renal vein involvement. Postoperatively, his glucose normalized, fevers resolved, and insulin was discontinued. Pathology confirmed the diagnosis of pheochromocytoma.
Discussion: Pheochromocytomas are rare tumors of adrenal chromaffin cells that secrete excess catecholamines, leading to variable presentations. This case illustrates the challenges in diagnosing pheochromocytoma, when presentations deviate from the classic triad of headache, palpitations, and diaphoresis. The patient’s severe hyperglycemia, intermittent fevers, and weight loss initially raised suspicion for infection or malignancy. However, labile severe hypertension alternating with orthostatic hypotension, combined with the imaging findings, pointed toward pheochromocytoma. The diagnosis was confirmed through biochemical tests showing elevated catecholamine metabolites and imaging results indicating an adrenal mass. Catecholamine release in pheochromocytomas is triggered by changes in tumor pressure or blood flow, explaining the patient’s hypertensive episodes and orthostatic hypotension. Hyperglycemia is thought to result from suppressed insulin secretion, increased glucagon release, and peripheral insulin resistance. Fevers in pheochromocytoma may arise from catecholamine-induced cytokine release, including IL-6, though other occult infections must be ruled out. Following tumor resection, catecholamine release ceases, leading to resolution of hyperglycemia, fevers, and other systemic effects.
Conclusions: This case underscores the importance of considering pheochromocytoma as a differential diagnosis for patients with atypical symptoms, particularly when unexplained hyperglycemia, intermittent fevers, and unintentional weight loss are present. Early diagnosis is crucial, as pheochromocytomas carry significant morbidity and mortality if left untreated.