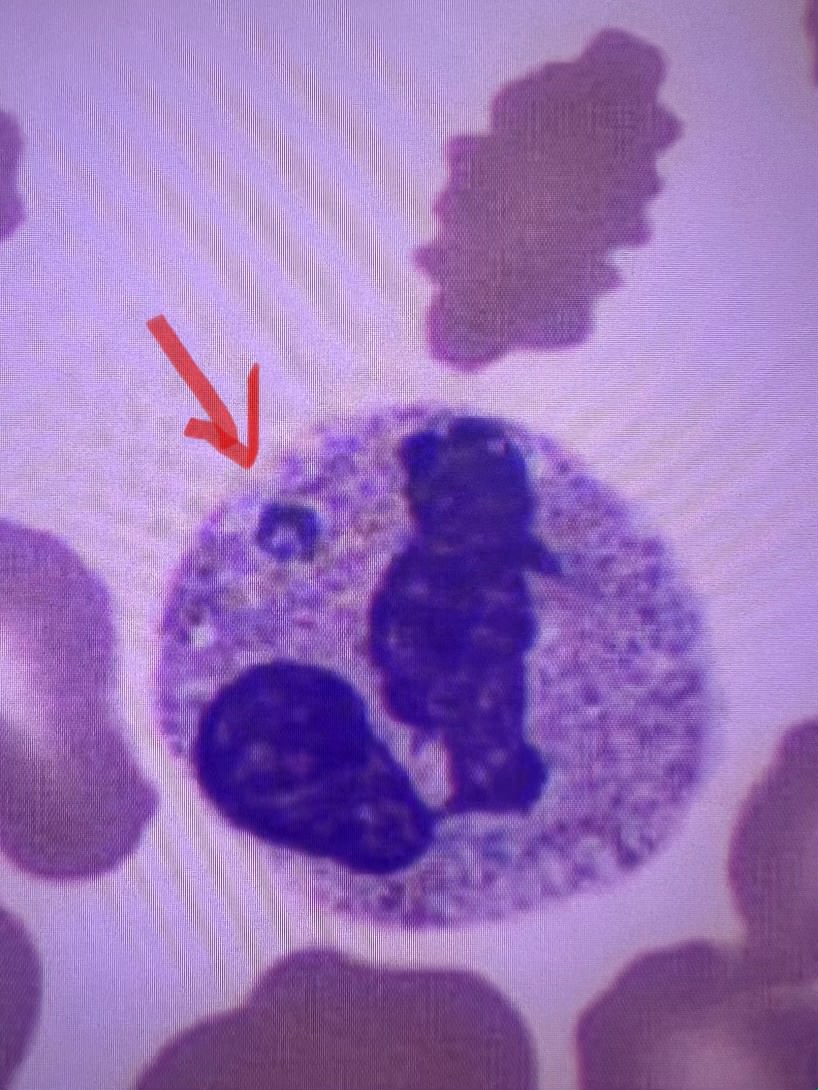
Case Presentation: An 87-year-old man with a history of chronic inflammatory demyelinating polyneuropathy (CIDP) on prednisone (7.5 mg daily), coronary artery disease post-CABG with pacemaker, Parkinson’s disease, COPD, and partial vocal cord paralysis presented with progressive weakness and pancytopenia. Over three weeks, he developed worsening asymmetric weakness, slurred speech, dysphagia, and high-grade fevers unresponsive to initial treatment for a presumed UTI.Upon transfer, labs revealed pancytopenia, severe hyperferritinemia (ferritin 15,768 ng/mL), elevated inflammatory markers, and liver enzyme abnormalities (AST 334 U/L, ALT 134 U/L). Imaging showed right lung opacities. His H-score of 264 strongly suggested hemophagocytic lymphohistiocytosis (HLH). Elevated soluble IL-2 receptor and CXCL9 levels, along with peripheral smear findings of intracytoplasmic inclusions, confirmed HLH secondary to anaplasmosis. He was treated with a 10-day course of doxycycline and tapered Anakinra, leading to resolution of HLH markers.Differential diagnoses for weakness included CIDP exacerbation, neuroborreliosis, neuroanaplasmosis, and tick-borne paralysis. Autoantibody testing (ACHR, MUSK, RNP, SCL70, HMGCR) was negative, with no evidence of myositis or Sjögren’s syndrome despite weakly positive SSA antibodies. B12 and folate were normal. Dysphagia improved with IV methylprednisolone, and he resumed maintenance prednisone. An outpatient EMG/NCS was arranged to further evaluate persistent weakness.
Discussion: HLH (hemophagocytic lymphohistiocytosis) is a syndrome marked by excessive immune activation, presenting with fever, organomegaly, cytopenias, and a significant increase in pro-inflammatory cytokines. While secondary HLH can be triggered by various infectious agents, Epstein-Barr virus is a common culprit, but anaplasmosis is a rare cause, particularly in elderly, immunosuppressed patients. This case is noteworthy due to several factors. First, HLH is more frequently diagnosed in pediatric populations or younger adults, with familial genetic causes being more common in children. In elderly patients, it is typically secondary to infections or malignancies, and chronic immunosuppression, as seen in this patient, likely contributed to the development of HLH. Second, the patient’s initial presentation, which included pancytopenia, neurological symptoms, and progressive weakness, raised concerns for other conditions such as stroke or myasthenia gravis exacerbation. The eventual diagnosis of HLH required a high degree of clinical suspicion, particularly with the presence of prolonged fever and multiorgan involvement. Lastly, while anaplasmosis generally causes a mild, self-limiting illness in healthy individuals, it can lead to severe disease in immunosuppressed patients. The pathophysiology of HLH triggered by anaplasmosis remains unclear but is thought to involve dysregulated immune responses.
Conclusions: This case underscores the importance of considering HLH in elderly patients with fever, pancytopenia, and multiorgan dysfunction, particularly when immunosuppression is present. Anaplasmosis should be included in the differential diagnosis of secondary HLH, especially in tick-endemic regions. Early diagnosis and treatment are critical to improving outcomes in these patients.