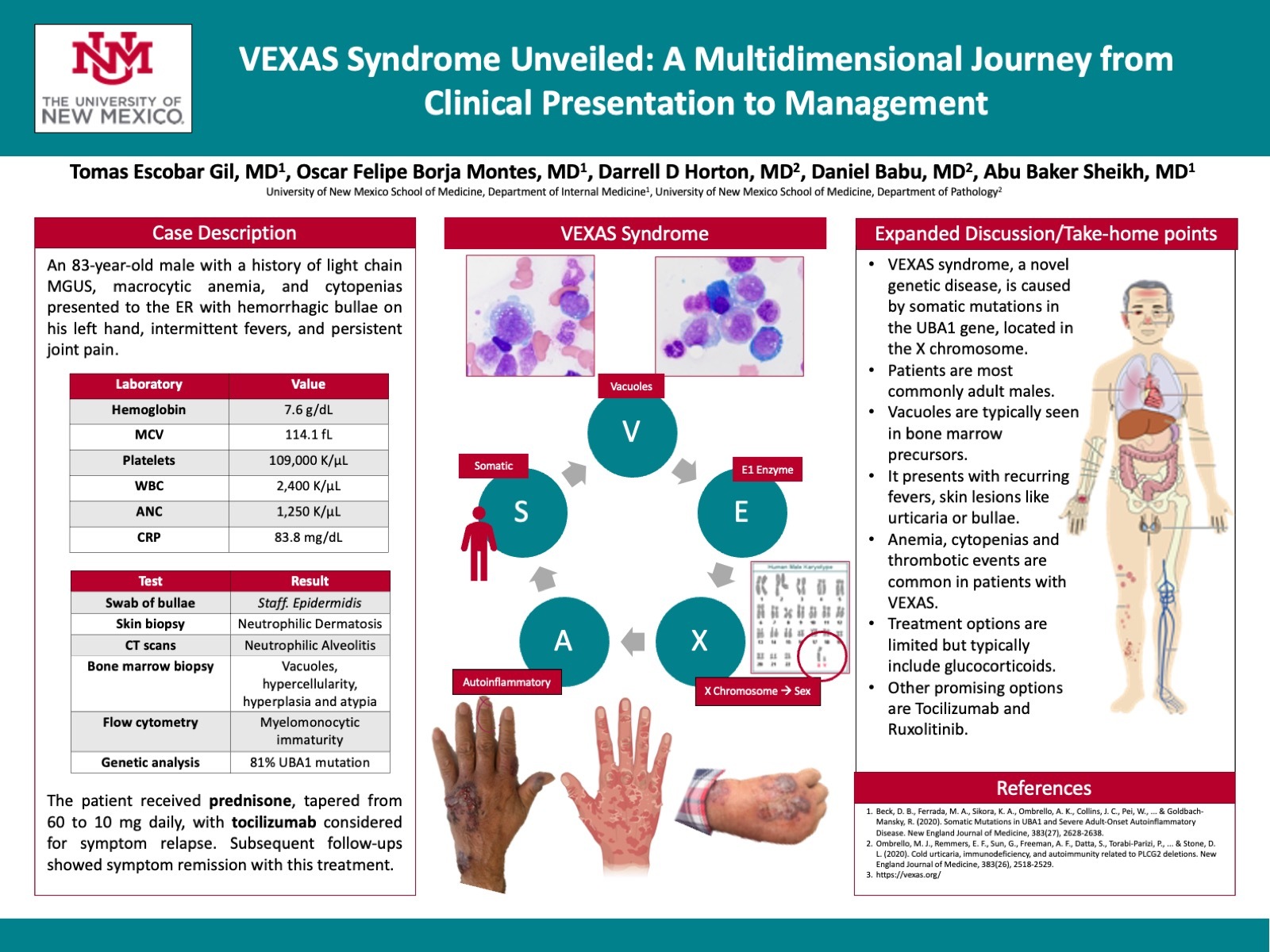
Case Presentation: An 83-year-old male with a past medical history of light chain MGUS, macrocytic anemia, and cytopenias, arrived at the emergency room presenting with hemorrhagic bullae on his left hand. He reported intermittent fever episodes and chronic, nonspecific joint pain. Admission labs indicated hemoglobin of 7.6, MCV of 114.1, platelet count of 109, and WBC count of 2.4 (ANC of 1.25). His CRP level was significantly elevated at 83.8. Initial treatment addressed a superficial skin infection of the hemorrhagic bullae, which yielded Staphylococcus epidermidis from a bacterial swab.A punch biopsy of the bulla identified a diffuse neutrophilic dermal infiltrate, scattered histiocyte cells, and dermal hemorrhage throughout the superficial and deep dermis, consistent with neutrophilic dermatosis. CT imaging revealed patchy multifocal opacities and clustered nodularity in the lungs, as well as mediastinal lymphadenopathy – findings suggestive of neutrophilic alveolitis. Subsequent bone marrow biopsy revealed hypercellularity (60%), pronounced myeloid and histiocytic hyperplasia, and an atypical immune cell population. Notably, vacuoles were observed in granulocytic and erythroid precursors, suggesting a clonal myeloid process. Flow cytometry identified 3% blasts with an immature myelomonocytic immunophenotype, expressing a range of CD markers and surface light chains. Cytogenetics presented a normal male karyotype, and a UBA1 mutation was identified (81%).These findings were deemed consistent with VEXAS syndrome. The patient commenced a regimen of prednisone, tapering from an initial dose of 60 mg daily to 10 mg daily, with potential consideration of tocilizumab as a steroid-sparing option upon symptom relapse. Subsequent follow-ups have indicated symptom remission under this treatment regime.
Discussion: VEXAS syndrome, a novel genetic disease resulting from UBA1 mutations, is characterized by inflammatory and hematologic symptoms due to alterations in UBA1 expression. The disease manifests clinically in varied ways, including systemic vasculitis and notable hematologic abnormalities. These patients presented with refractory constitutional symptoms, ear and nose chondritis, inflammatory arthritis, vasculitis, elevated inflammatory markers, macrocytic anemia, thrombocytopenia, dysplastic bone marrow findings with vacuoles in myeloid and erythroid precursors. Notably, venous thromboembolism can occur spontaneously, suggesting an enhanced pro-thrombotic tendency in these patients. Currently, treatment options are limited, with glucocorticoids offering some symptomatic relief, and ruxolitinib showing potential as a more effective therapeutic approach. Clinicians need to maintain a high suspicion for VEXAS syndrome in patients with unexplained inflammatory and hematologic symptoms and consider UBA1 mutation testing.
Conclusions: In summary, our case highlights the complex nature of VEXAS syndrome. With its diverse clinical profile and limited treatment options, this condition poses challenges for both clinicians and patients. The diagnostic journey of our patient, characterized by a comprehensive workup and a positive response to glucocorticoids, speaks to the need to approach VEXAS cases on a case-to-case basis. As we look to the future, emerging treatments like Ruxolitinib offer hope, but further research is needed to define the optimal management strategy.