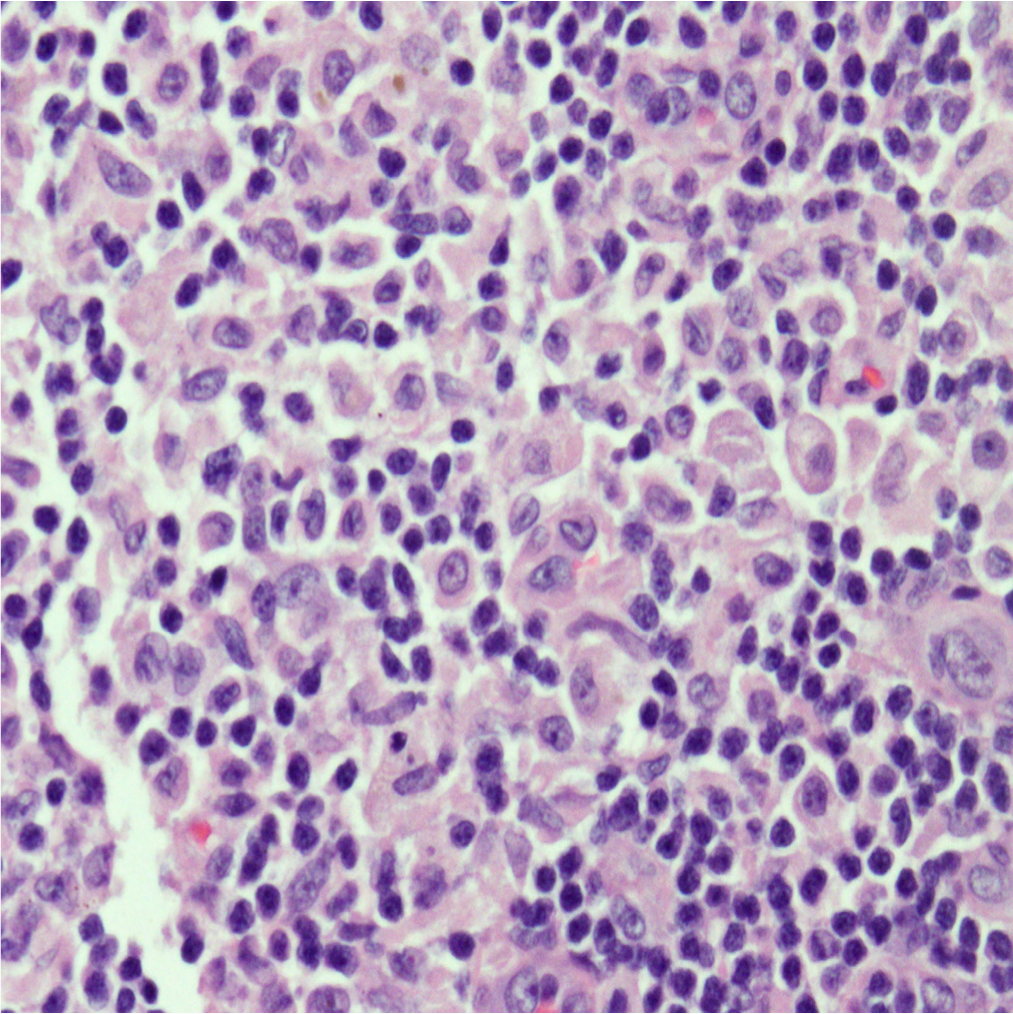
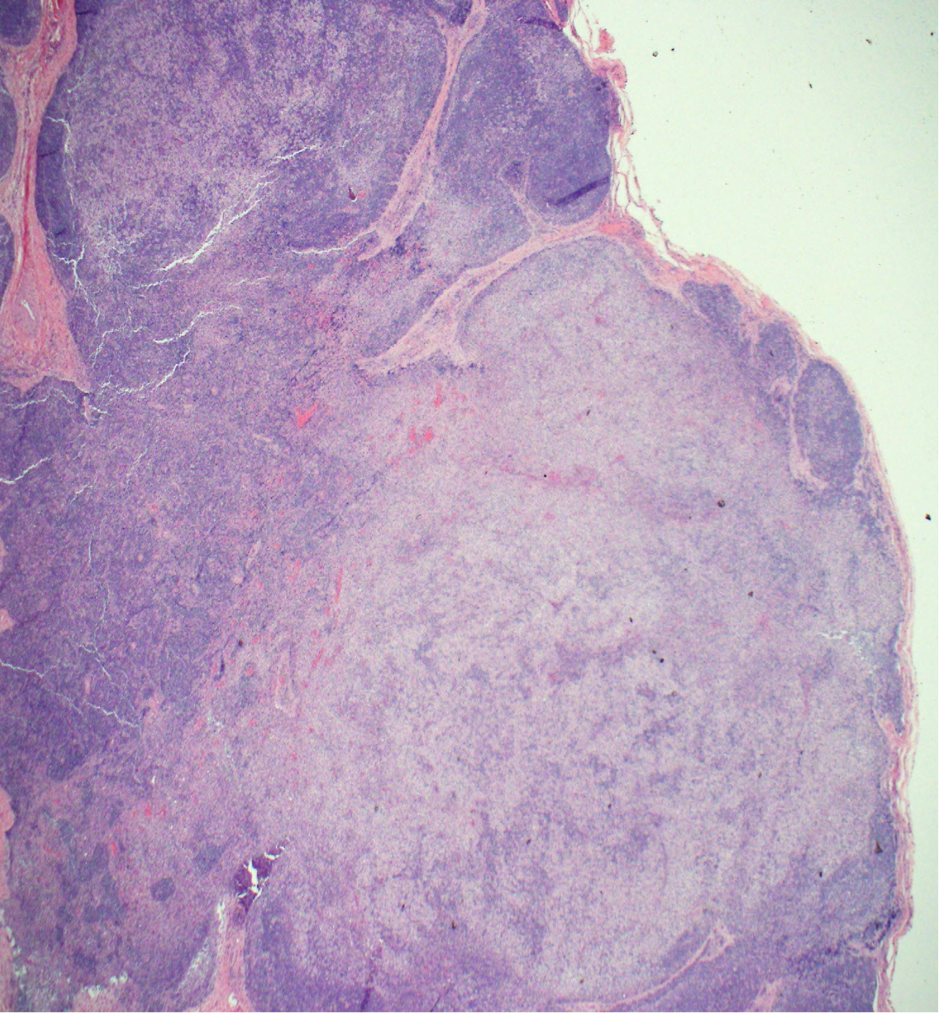
Case Presentation: A 21 year old man presented to his primary care doctor with 4 days of fever and pruritic rash on his hands and feet. Labs showed leukocytosis and transaminitis. He was started on doxycycline for Rocky Mountain Spotted Fever, however tick borne serologies, MonoSpot, HIV, RPR and hepatidities were ultimately negative. He presented to the hospital 4 days later with cough and night sweats. Exam revealed a thin man with cervical and axillary lymphadenopathy, an erythematous rash on his extremities, pedal edema, and petechiae on his bilateral feet. Labs showed worsening transaminitis and eosinophilia. Chest CT showed mediastinal and hilar lymphadenopathy with pneumonitis. Infectious work up for bacterial, fungal, and viral pathogens was negative. Rheumatologic studies were also unremarkable. Flow cytometry showed no B-cell or T-cell abnormalities. He lost 25 pounds during admission and developed thyroiditis and anterior uveitis. Axillary lymph node biopsy revealed cells staining positive for CD1a, S100, CD68, and langerin (CD207) consistent with Langerhans cell histiocytosis (LCH). Treatment with vinblastine and prednisone was initiated with resolution of his symptoms and normalization of his labs.
Discussion: LCH is a rare disorder characterized by clonal proliferation of abnormal dendritic cells with varying presentations ranging from single-organ involvement to disseminated disease. It is more common in children 1 to 3 years old but can be seen in all age groups. The majority of patients have lytic bone lesions and about 30% have skin involvement. LCH has been historically separated from other malignancies, however treatment involves both steroids and chemotherapy.
Diagnosis is tricky as many different organs may be affected and it can resemble more common disorders. It is based on pathologic evaluation of involved tissue and correlation with clinical presentation. No cytogenetic findings are diagnostic, but obtaining a tissue sample (bone, skin, lymph node) in these patients can help shed light on the underlying disease process and lead to earlier diagnosis and treatment.
Conclusions: LCH is a rare histiocytic disorder with a wide range of presentations that can present at any age. Because symptoms of LCH are variable and nonspecific and more commonly diagnosed during childhood, adults presenting with LCH frequently go weeks to months before they are diagnosed. Physicians must have a high degree of suspicion in patients with fevers, eosinophilia, rash, and lymphadenopathy to accurately diagnose their patients. Clinical trials provide opportunities for better knowledge of LCH treatment in adults. Patients should undergo post-treatment surveillance to monitor for signs of possible relapse and long-term effects of therapy.