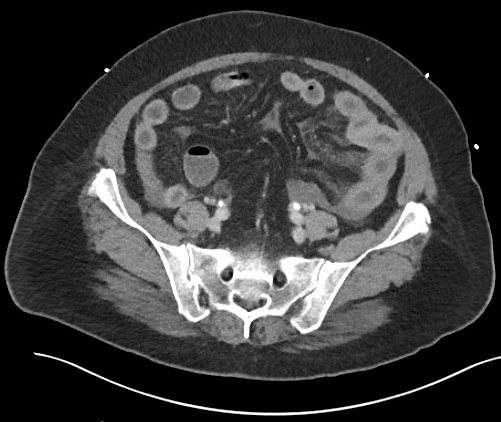
Case Presentation: Our patient is a 58 year-old Portuguese-speaking female with a history of CLL and autoimmune hemolytic anemia who presented with severe epigastric pain, nausea, and vomiting. She presented with similar symptoms requiring two prior admissions within the last 6 weeks. CT scan of the abdomen and pelvis showed bowel wall thickening and hyperenhancement of the jejunum with mesenteric engorgement. Stool Carey-Blair and Clostridium difficile PCR testing were negative; her symptoms improved over one to two days, and were attributed to viral gastroenteritis. She presented four weeks later with similar symptoms. Repeat CT scan showed persistent, but improved inflammatory change involving the jejunum. She was again diagnosed with viral gastroenteritis, treated with supportive care and had subsequent improvement of symptoms within 24 hours. A hepatobiliary iminodiacetic acid (HIDA) scan was normal and outpatient colonoscopy was arranged.
Four days after being discharged, she presented once again with similar symptoms. A small bowel follow through during this admission was normal. Upon review of her medical history, her recurrent abdominal pain, nausea, and vomiting began shortly after completing a prednisone taper to treat autoimmune hemolytic anemia related to her CLL (diagnosed nearly six months prior to this presentation). She had been started on prednisone 90 mg daily for one week, then tapered to 20 mg daily and weaned off steroid therapy three weeks before her first hospitalization for these recurrent symptoms. Our differential at this time included Acquired Angioedema due to deficiency of C1 esterase inhibitor C1INH-AAE as a complication from CLL vs. small bowel vasculitis vs. inflammatory bowel disease. Laboratory results showed very low C4 (<3), normal C3 (102), normal C1q binding (<1.2), and very low functional C1 esterase inhibitor (15%). She was recently started on Rituximab for treatment.
Discussion: This case illustrates the difficulty in recognizing acquired small bowel angioedema secondary to C1 esterase inhibitor deficiency. Given her history of CLL and recent episode of autoimmune hemolytic anemia, our patient had two underlying, albeit concomitant, risk factors for C1INH-AAE.3 She required recurrent hospital admissions with extensive workup, including multiple laboratory studies and imaging series contributing significantly to healthcare expenditures, before the diagnosis was elucidated. Consideration of this potential differential is key in accurately working up the patient with recurrent, unexplained abdominal pain, diarrhea, and nausea/vomiting with history of lymphoproliferative or autoimmune phenomena, and especially in patients with both conditions.
Conclusions: In patients with recurrent abdominal symptoms and recurrent episodes of angioedema associated with B cell lymphoproliferative disorders and autoimmunity the diagnosis of C1INH-AAE should be considered.