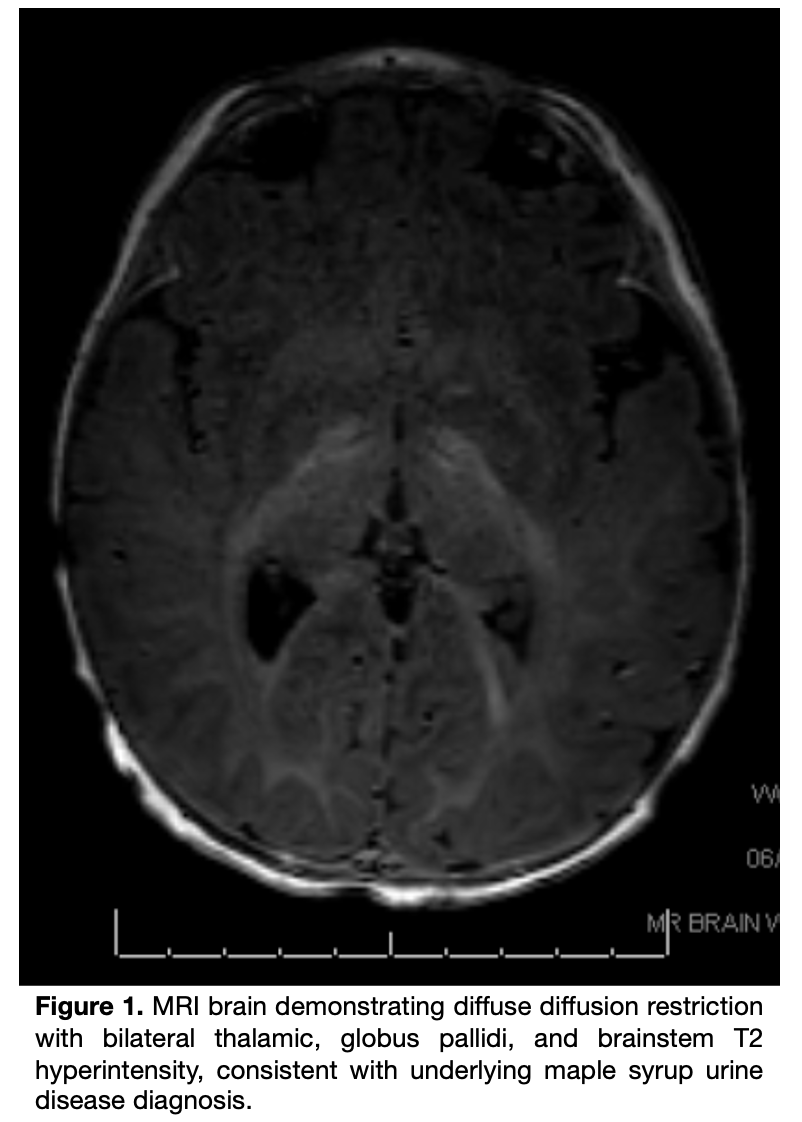
Case Presentation: A 9-month-old unvaccinated female with no past medical history presented with three days of non-bilious, non-bloody vomiting after contact with a sick child. Parents denied fever, diarrhea, or rash. Initially vomiting after each feed, she progressed to near-hourly emesis, became lethargic, and voided only once on the day of admission. The child had never been evaluated by a physician because her parents did not believe in the medical system. In the emergency department, she appeared ill, somnolent, and dehydrated. Laboratory evaluation revealed elevated anion gap metabolic acidosis with undetectable bicarbonate and ketonuria, initially presumed to represent gastroenteritis. She required ICU admission for fluid resuscitation; although acidosis corrected, she remained unable to tolerate oral intake but was transferred to the acute care floor. Upon arrival, she was somnolent with recurrent emesis, hepatomegaly, tremulous upper extremities, hypotonia, and pallor. Given her mental status, she was transferred back to the ICU. Further laboratory work-up revealed normal ammonia, electrolytes and glucose, low bicarbonate of 17 mmol/L, and mild transaminase elevation. An MRI brain obtained due to altered mental status showed diffuse diffusion restriction with bilateral thalamic, globus pallidi, and brainstem T2 hyperintensity (Fig. 1). Plasma amino acids revealed elevated valine, leucine, and isoleucine. Urine organic acids showed ketosis with elevated branched-chain ketoacids. Genetic testing confirmed two pathogenic DBT variants, establishing the diagnosis of Maple Syrup Urine Disease (MSUD).
Discussion: MSUD is a rare autosomal recessive disorder caused by deficiency of the branched-chain α-ketoacid dehydrogenase complex, impairing breakdown of leucine, isoleucine, and valine. Catabolic stress from illness or fasting leads to toxic metabolite accumulation, neurotoxicity, and cerebral edema. Anchoring on a diagnosis of gastroenteritis in a pediatric patient with isolated vomiting can delay recognition of serious underlying metabolic or neurologic disease. While gastroenteritis is common, it typically presents with accompanying diarrhea and low-grade fever; isolated or progressive vomiting without these features should prompt broader consideration, including inborn errors of metabolism, intracranial pathology, or toxic ingestion. Avoiding premature diagnostic closure is particularly important in infants and unvaccinated or medically unmonitored children, where subtle signs may represent life-threatening pathology rather than benign illness. This patient’s delayed diagnosis reflects both the absence of newborn screening and delayed medical care because her parents did not believe in the medical system. Her later presentation is consistent with intermittent MSUD, a milder phenotype compared to classic neonatal-onset disease.
Conclusions: This case underscores two key lessons: (1) seemingly common presentations, such as vomiting presumed to be gastroenteritis, require diagnostic vigilance, as rare metabolic disorders can present in a similar manner; and (2) systemic barriers, including lack of newborn screening and delayed medical evaluation due to parental mistrust, can postpone recognition of life-threatening disease. Early suspicion and timely intervention are essential to prevent irreversible neurologic injury and improve outcomes in children with undiagnosed metabolic disorders.