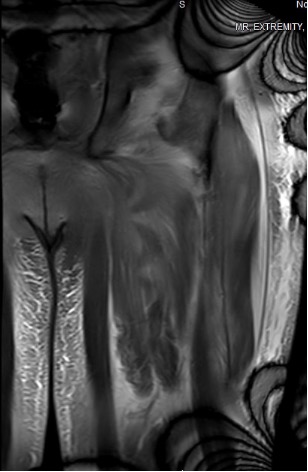
Case Presentation: A 61-year-old male with past medical history of hypertension presented for three months of proximal muscle weakness. He had progressive weakness in his legs and shoulders to the point of wheelchair dependence. He also reported dysphagia to solids for two weeks. Workup was notable for anti-nuclear antibody (ANA) 1:1280, elevated aldolase levels, and positive anti-signal recognition particle (anti-SRP) antibody. MRI of the lower extremities showed symmetric diffuse edema and moderate atrophy of the bilateral pelvic girdle and thigh muscles particularly involving the adductor compartment muscles with fascial edema, most compatible with chronic inflammatory myositis (Figure 1). CT of the chest, abdomen, and pelvis was negative for any primary or metastatic disease but showed fluid within the intramuscular planes of the right lateral abdominal wall and anterior compartments of the thighs, possibly representing myositis. He was empirically treated with intravenous immunoglobulin (IVIG) for five days. He underwent biopsy of his right deltoid and left quadriceps muscle. His muscle biopsy pathology results were positive for MHC-I, which confirmed immune-mediated necrotizing myopathy. He was discharged with rheumatology follow-up with plans to continue to receive IVIG every four weeks.
Discussion: Inflammatory myopathies are characterized by proximal muscle weakness and elevated creatine kinase levels [1, 2]. Immune-mediated necrotizing myopathy (IMNM) is distinguished from other idiopathic inflammatory myopathies (IIMs) such as dermatomyositis in that biopsy shows minimal lymphocytic infiltrates and necrotic tissue [2]. Those with IMNM may have positivity to the signal recognition particle (SRP) antibody, hydroxy-3-methylglutaryl-CoA reductase (HMGCR) antibody, or neither. The anti-SRP subtype of IMNM is much rarer, making up approximately 20% of IMNM cases and 5% of IIM cases [3, 4]. We highlighted a rare case of anti-SRP positive IMNM here. Biopsy is indicated to diagnose IMNM when autoantibody testing is negative or antibody results are delayed. The MHC-I positivity on immunostaining is known to be a sensitive marker for diagnosis of IMNM [5]. On the other hand, positive MHC-II immunostaining can help differentiate IMNM and other IIMs from other hereditary myopathies [6, 7]. The standard treatment of IMNM includes high-dose steroids combined with steroid-sparing agents such as methotrexate. Those with refractory disease should receive IVIG or rituximab. Our patient was initiated directly on empiric IVIG due to poor prognostic factors including anti-SRP-positive disease, presence of muscle atrophy on MRI imaging, and severely elevated creatine kinase levels. Cardiac involvement including acute coronary syndrome and arrhythmias has been reported in up to 30% to 40% of patients in multiple studies as well. Our patient was found to have elevated troponin levels but normal electrocardiograms without ischemic changes, so further cardiac imaging was not pursued.
Conclusions: The standard treatment for IMNM is steroids followed by IVIG or rituximab for steroid-refractory cases. However, clinicians should consider starting with IVIG or rituximab for statin-naive anti-HMGCR and anti-SRP subgroups as these patients have been shown to have steroid-refractory disease. Given the prevalence of cardiac manifestations in IMNM, screening for cardiac abnormalities should be a keystone in IMNM patients and can improve patient outcomes.