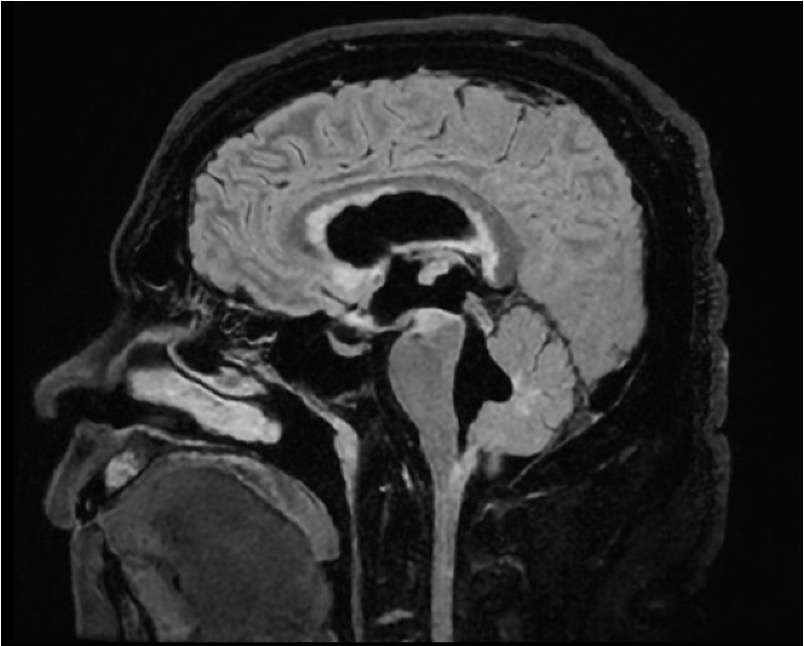
Case Presentation: A 42-year-old African American male was admitted with nausea, vomiting, dizziness, and abdominal pain after a recent cholecystectomy. He was afebrile, with blood pressure 117/67 mmHg, respiratory rate 20, and oxygen saturation 99%. Initial workup included CT Chest, Abdomen, and Pelvis; relevant for a small pneumoperitoneum attributed to postoperative status. Despite abdominal pain resolution, nausea, vomiting, and vertigo continued. Further physical exam revealed bidirectional nystagmus and papilledema, without anterior uveitis. Brain MRI brain revealed severe communicating hydrocephalus and leptomeningeal enhancement. On the same day, he became febrile, and meningitis treatment was initiated. Lumbar puncture showed an elevated opening pressure of 38 cm H2O, WBC 120 (91% lymphocytes), RBC 28, elevated protein 138, low glucose 33, and negative flow cytometry. MRI of the spine showed diffuse nodular enhancement. After a negative infectious workup, treatment for presumptive neurosarcoidosis was started: 60 mg of prednisone daily and cyclophosphamide. Follow-up lumbar puncture showed persistently elevated opening pressure, and a ventriculoperitoneal shunt was placed. Leptomeningeal biopsy showed dystrophic calcifications. Initial symptoms resolved, and patient was discharged on a prednisone taper, monthly cyclophosphamide infusions, and close multidisciplinary follow-up.
Discussion: Sarcoidosis is a multisystem inflammatory disorder defined by epithelioid non-caseating granulomas, with a typical age at diagnosis of 35-50 years old, occurring more frequently in African Americans and females (1). Neurosarcoidosis (NS) is a rare manifestation, affecting 3-10% of patients (2). Less than 1% of patients with NS lack systemic sarcoidosis (3). Clinical symptoms include optic neuritis, intraparenchymal lesions, seizures, depression, spinal cord involvement, hydrocephalus, and peripheral neuropathy (2,4). Given that NS can mimic several disease processes, extensive workup is required to confirm the diagnosis. Lumbar punctures often show elevated protein and lymphocytosis (5). Use of CSF ACE and serum ACE has been debated due to poor diagnostic performance. Notably, both were ordered and positive in our case. Brain MRI often shows leptomeningeal involvement (4). The gold standard for diagnosis is a biopsy with non-caseating granulomas, although it can often be negative, with a sensitivity of 65% (6). Our patient’s biopsy failed to show granulomas, although it helped rule out malignancy and infection. Suspected cases are labeled as possible, probable, or definitive NS (8). Our patient met the “possible” criteria due to the lack of granulomatous pathology. Corroborating laboratory findings, brain biopsy ruling out secondary causes, and response to treatment build the ultimate case for NS in some patients. Treatment relies on glucocorticoids and immunosuppressants, although randomized clinical data are lacking (2,6). Although steroids can initially be effective, patients often require immunosuppressive therapy (9,10). Unfortunately, treatment remains challenging, and up to one-third of patients fail to respond (10). Although the initial response in our patient is promising, close multidisciplinary follow-up is required.
Conclusions: Neurosarcoidosis is a rare manifestation of sarcoidosis that poses significant diagnostic challenges. Prompt treatment with steroids and often immunosuppressants is imperative to prevent irreversible damage.