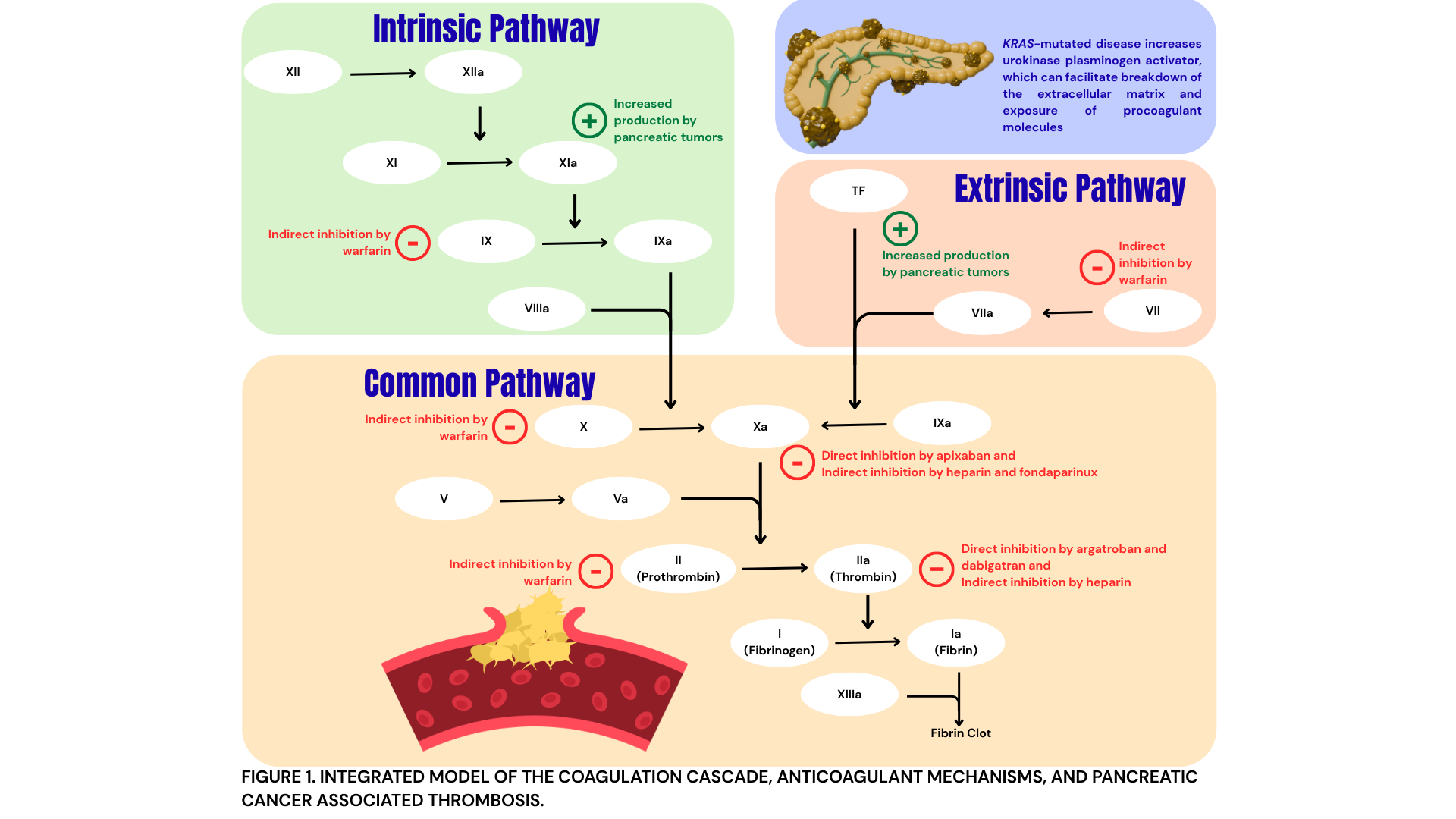
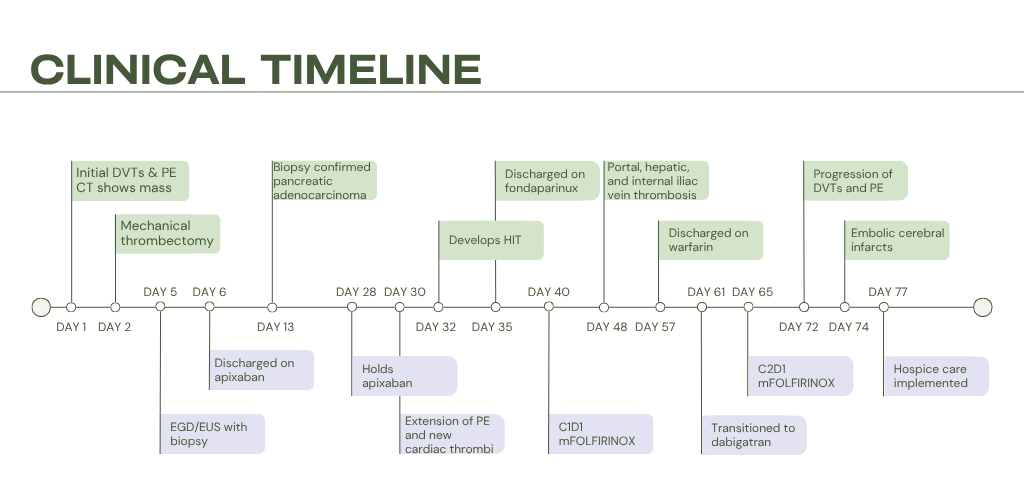
Case Presentation: A 67-year-old female with a history of hypertension, dyslipidemia, and type 2 diabetes mellitus presented with pleuritic chest pain, palpitations, and dyspnea. She reported a 24-lb intentional weight loss, but no cough, hemoptysis, trauma, fever, or travel. She denied tobacco use. An electrocardiogram showed sinus tachycardia; physical examination was otherwise unremarkable. Labs were normal aside from elevated alkaline phosphatase (253 U/L) and pro-brain natriuretic peptide (1125 pg/mL). Computed tomography (CT) pulmonary angiogram revealed a pulmonary embolism (PE) with bilateral lobar branch involvement and multiple hepatic lesions. Venous duplex showed bilateral lower-extremity deep vein thromboses (DVTs). Transthoracic echocardiogram showed right ventricular apex akinesis, representing right-sided heart strain. She was started on heparin and underwent thrombectomy. Abdominal CT revealed a 3.5 × 2.6-cm pancreatic-neck mass and bilobar hepatic lesions. Biopsy confirmed pancreatic adenocarcinoma with a KRAS G12D mutation, MYC amplification, CDK2NA/B loss, and microsatellite stability. She was discharged on apixaban. One week later, after holding three doses of apixaban for planned port placement, she developed worsening lower extremity swelling with extension of her DVTs and PE and formation of new intracardiac thrombi. Heparin was restarted, but her course was complicated by heparin-induced thrombocytopenia (HIT) confirmed by positive heparin-PF4 antibodies, prompting transition to argatroban and later fondaparinux. Weeks later, after initiation of chemotherapy, she developed recurrent, rapidly progressive thrombosis involving the portal, hepatic, and iliac veins. She was again transitioned to argatroban and bridged to warfarin, and later dabigatran due to unpredictable fluctuations in INR. Clot burden again progressed, necessitating resumption of argatroban, during which she suffered from multiple embolic cerebral infarcts. Despite multiple lines of anticoagulation, she experiences relentless thrombosis and clinical decline; ultimately, she elected hospice care and expired shortly after.
Discussion: This case illustrates the profound hypercoagulability of advanced pancreatic adenocarcinoma, especially KRAS-mutated disease. Pancreatic tumors increase thrombotic risk through production of tissue factor and other procoagulant factors, enhanced platelet and endothelial activation, and production of inflammatory mediators. KRAS mutations further increase risk by increased production of urokinase plasminogen activator. Cancer-associated thrombosis is a poor prognostic factor; in this patient, malignancy-driven coagulopathy progressed despite six anticoagulants and was complicated by HIT. Her course illustrates the limitations of current anticoagulation strategies in aggressive pancreatic cancer and highlights the need for better biomarkers to identify drivers of refractory thrombosis, improved risk stratification for HIT, and clearer guidance on selection of non-heparin agents. Early multidisciplinary involvement is essential to align treatment with patients’ goals and expectations.
Conclusions: Refractory cancer-associated thrombosis is of significant prognostic value and poses significant therapeutic and ethical challenges. Recognizing when recurrent thromboembolic events persist despite optimal therapy is critical to guiding timely transition from disease-directed treatment to comfort-focused care.