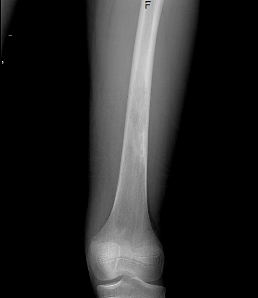
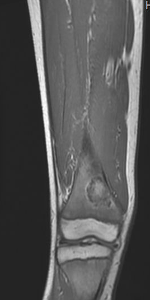
Case Presentation: A 12-year-old Caucasian male with no significant medical history presented with a 6-day history of bilateral leg pain following a soccer game. As an avid soccer player, he had experienced similar episodes over the past three years, managed previously as “growing pains” with NSAIDs and acetaminophen. However, this presentation raised concern for an underlying condition, considering the recurrent nature of the pain. The review of systems was negative for constitutional symptoms. Clinical evaluation revealed tenderness upon palpation of both distal femora. Laboratory investigations showed a normal complete blood count with mild microcytosis, Mentzer index of 13.09, normal electrolytes, and normal inflammatory markers. Radiographic examination revealed intriguing findings—a left femur radiograph displayed endosteal scalloping and patchy lucencies extending approximately 12.5 cm with ill-defined margins, and a skeletal survey revealed a well-demarcated, rounded, lucent lesion in the left distal femur metadiaphysis measuring approximately 17 x 15 mm. Magnetic resonance imaging (MRI) with contrast exhibited subperiosteal fluid collections along both femora and a localized hyperintensity with peripheral contrast enhancement in the right femur measuring 2.4 x 3.6 x 2 cm. Further analysis through hemoglobin electrophoresis revealed HbA 24.3%, HbA2 4.6%, HbF 8.6% and HbS 62.5%, thus an unexpected finding of HbS/Beta-thalassemia was made as workup for bone infarction and mild anemia.
Discussion: The differential diagnosis encompassed a spectrum of conditions, including hematological malignancies like acute lymphoblastic leukemia, bone cysts, masses, and bone infarcts. Intriguingly, the MRI findings were consistent with bone infarction, a manifestation of sickle cell disease (SCD) and beta-thalassemia. Osteonecrosis is a described complication of SCD in up to 10% of cases. Usually, epiphyses are involved. Vaso-occlusive episodes, hypercoagulability, marrow edema and expansion are contributing factors for these phenomena. In this specific case, the combination of pathologies predisposed the patient to higher risk. Despite the rarity of this presentation, the patient’s familial history, with the father being a carrier for sickle cell trait and the mother for beta-thalassemia minor and multiple miscarriages, guided the diagnosis. This information was not disclosed on the initial interview.
Conclusions: This case presents a diagnostic challenge in identifying concurrent hemoglobinopathies and bone lesions in pediatric patients. The recognition of subtle clinical cues, meticulous investigation, and collaboration among multiple specialties—pediatric hematology, radiology, and pediatric hospitalists—were instrumental in unraveling this complex diagnosis. It highlights the importance of expanding differential diagnoses to include rare but crucial conditions, emphasizing the need for comprehensive evaluations in recurring extremity pain cases. Such interdisciplinary approaches facilitate timely and accurate diagnoses, ensuring appropriate management strategies and improved patient outcomes.This abstract elaborates on the clinical findings, diagnostic journey, and emphasizes the significance of interdisciplinary collaboration in elucidating complex medical conditions in pediatric patients.