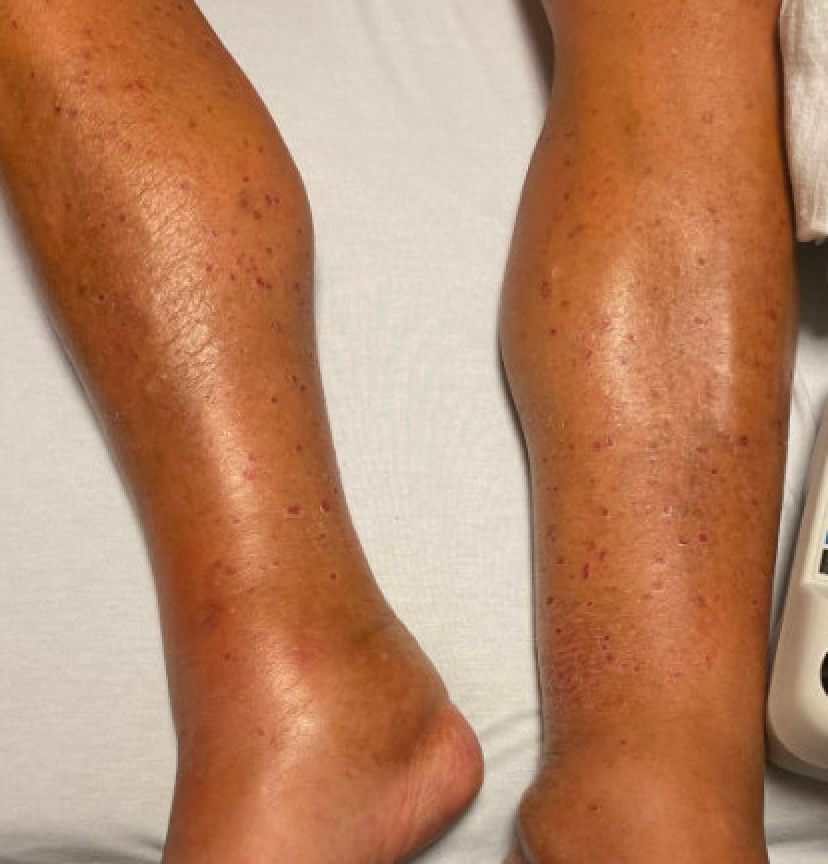
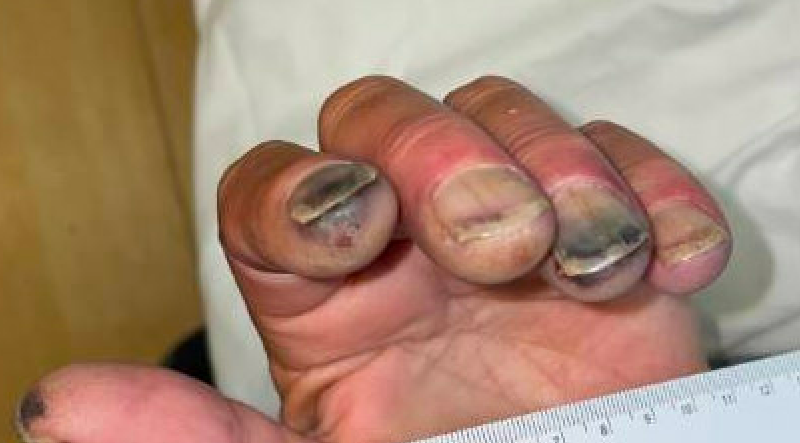
Case Presentation: A 61-year-old male with past medical history of type 2 diabetes, hypertension, hyperlipidemia, tobacco use disorder, Raynaud phenomenon, and provoked DVT presented from an outside hospital with abrupt onset of a purpuric lower extremity rash with discoloration and ulceration of the distal fingers and toes. At the time of the patient’s DVT several years prior, labs revealed the presence of anticardiolipin antibodies (anti-beta-2-glycoprotein antibody testing was not performed). He was transferred to our tertiary facility out of concern for catastrophic antiphospholipid syndrome. Treatment for endocarditis was initiated given exam findings compatible with showering emboli. Antiphospholipid antibodies were drawn, and the patient was found to be positive for anticardiolipin IgM (30.3 units/mL) and anti-beta-2-glycoprotein IgM (36.3 units/mL) antibodies. Transesophageal echocardiogram and blood cultures were negative. CTA thorax and CTA abdomen/pelvis with runoff were significant only for short segment, non-flow-limiting left popliteal artery dissection. Bilateral lower extremity USV with Doppler demonstrated a long segment of left small saphenous vein thrombus but was negative for DVT. MRI/MRA/MRV brain were unremarkable. ABIs were within normal limits. Ultimately, skin biopsy of the right hallux was performed which was consistent with leukocytoclastic vasculitis with vascular thrombosis and overlying epidermal ulceration and necrosis.
Discussion: Given the detection of anticardiolipin antibodies on two separate occasions, a diagnosis of antiphospholipid syndrome (APS) was considered. However, antibody titers were not perceived to be elevated to a level that would explain the patient’s degree of ischemia. In addition, the vessel wall inflammation that was seen on histopathology in our patient is not typically seen in APS. Endocarditis can certainly cause embolic ischemia, but the patient’s negative echocardiogram and blood cultures rendered this unlikely, and antibiotics were stopped on transfer. Taking into account his history of smoking and overall clinical presentation, histopathologic findings were thought to align with a diagnosis of thromboangiitis obliterans (TAO). TAO is an inflammatory vasculitis with classic arteriographic findings of nonatherosclerotic segmental occlusions of small and medium arteries. TAO is most frequently diagnosed in males who smoke and is thought to involve components of tobacco hypersensitivity, autoimmunity, and genetic predisposition. Though commonly used diagnostic criteria specify an onset of symptoms before the age of 45 to 50 years, some data suggest that a proportion of patients with TAO may present at an older age. Generally a diagnosis of exclusion, treatment for TAO involves calcium channel blockers for symptom management and smoking cessation.
Conclusions: This patient’s case is unique in that he presented with sequelae that can be seen in both TAO and APS. His age also fell outside of the range typically associated with TAO. This case highlights the concept that though these conditions are two distinct clinical entities, TAO and APS can co-occur and both diagnoses should be considered in a patient presenting with peripheral occlusive vascular disease. In fact, there is emerging evidence that anticardiolipin antibodies can be associated with TAO and may contribute to the severity of thrombosis. In addition, age outside of the usual expected range should not preclude consideration of TAO in the differential diagnosis.