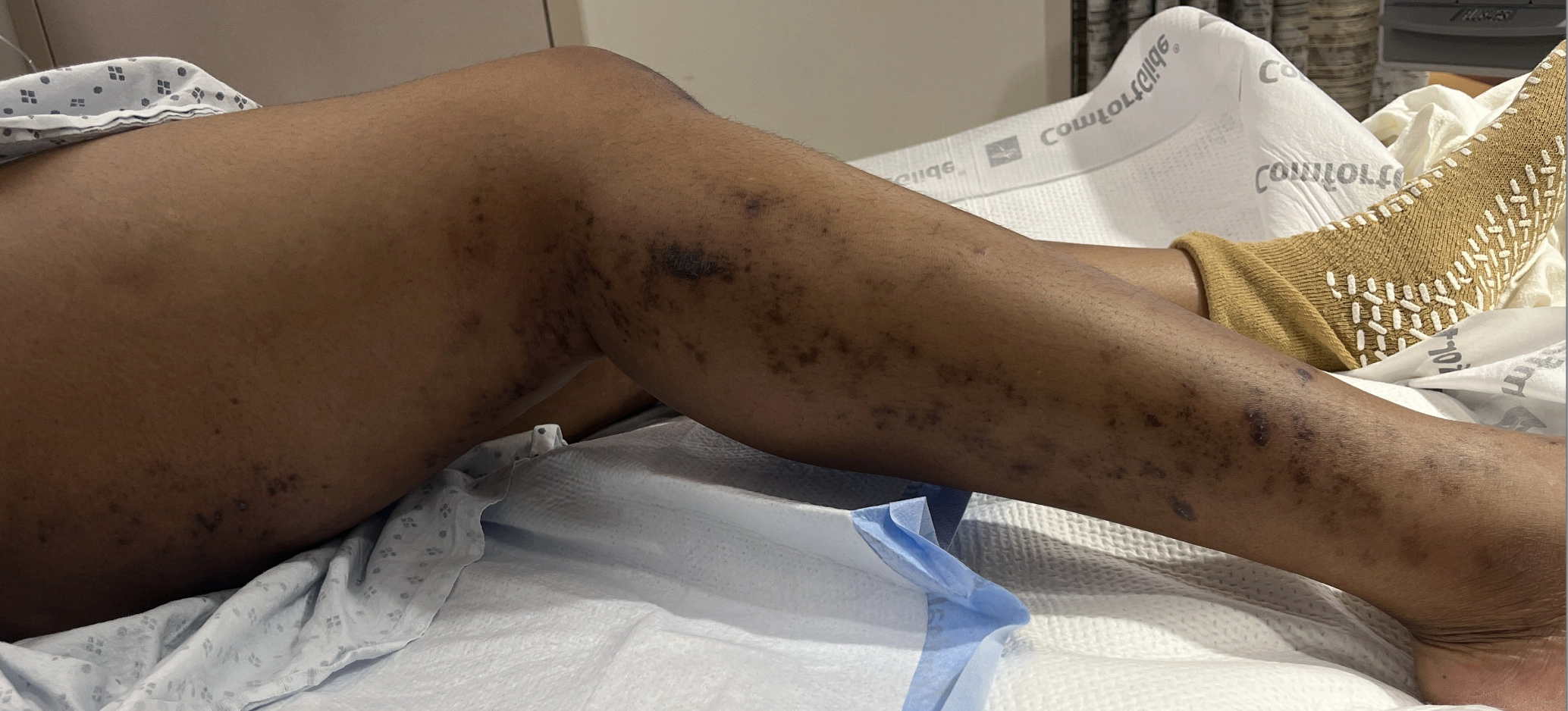
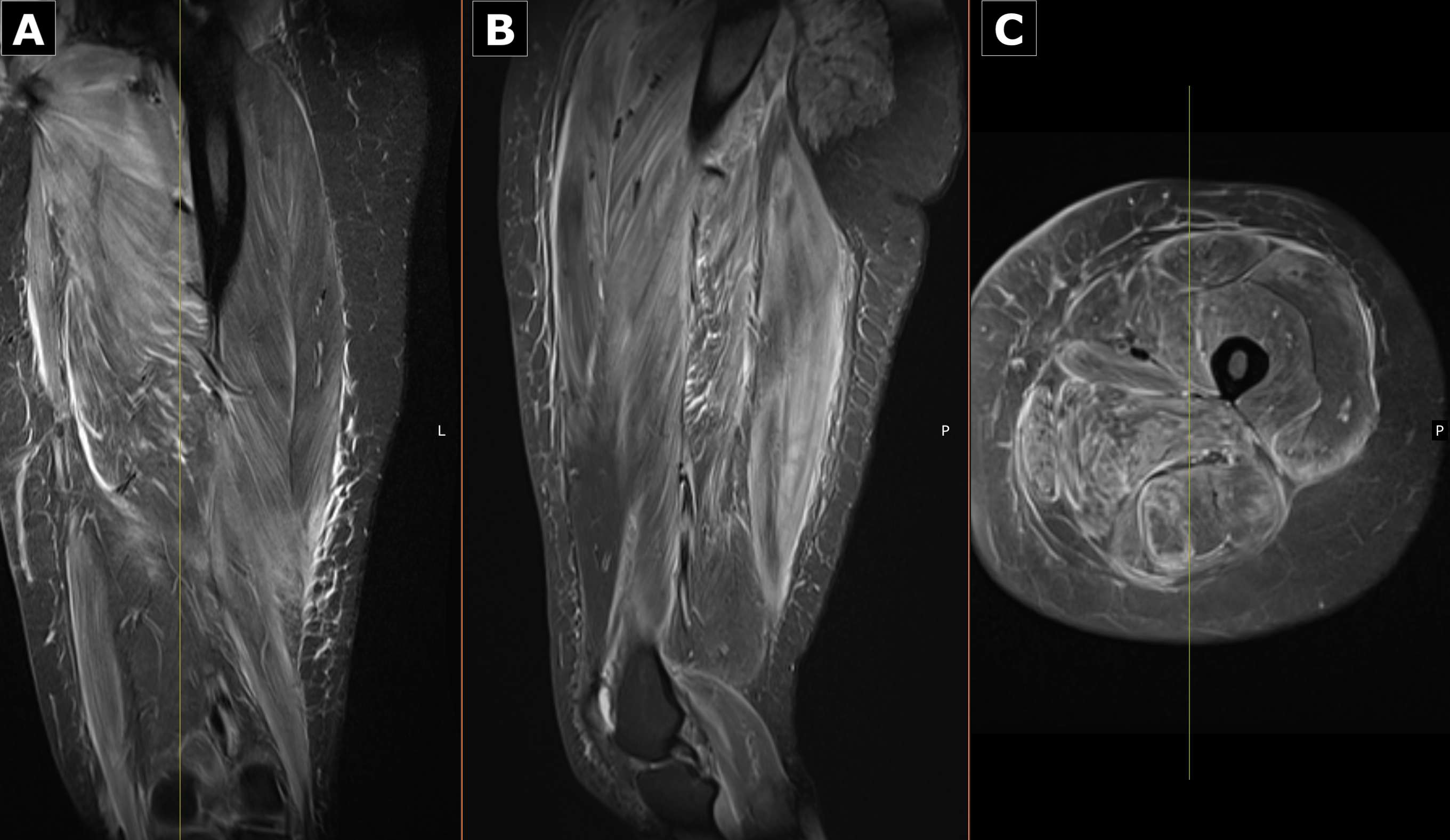
Case Presentation: A previously healthy 30-year-old woman presented with progressive myalgia, intermittent fevers, and worsening proximal weakness. She immigrated from Western Africa five months earlier with a remote history of malaria. For nearly a decade, she reported intermittent mild myalgias and exertional cramps that slowly advanced to significant functional limitation. She denied recent illness, surgery, medication changes, or vaccinations. Her only medication was a prior depot medroxyprogesterone injection, and there was no family history of hepatic, autoimmune, or neuromuscular disease.On arrival, she was febrile to 101.3 °F and tachycardic, with diffuse muscle tenderness that markedly limited active movement; passive motion remained intact. Trace edema was present, and she had a hyperpigmented, non-vesicular papular rash extending from mid-thigh to the foot. Neurologic examination remained nonfocal.Laboratory evaluation revealed striking elevations in CK (31,103 U/L), AST (1,025 U/L), ALT (868 U/L), aldolase (338.6 U/L), LDH (1,809 U/L), and TSH (10.5 μIU/mL). ESR was 42 mm/hr and CRP 3.6 mg/dL. Urinalysis showed myoglobinuria.Given the combination of fever, weakness, and transaminitis, infectious and autoimmune causes were investigated. She briefly received artemether–lumefantrine and doxycycline, discontinued after negative studies. An initial Monospot was not supported by EBV serologies.Persistent weakness prompted an MRI of the left thigh, demonstrating diffuse intramuscular edema and enhancement concerning for inflammatory myopathy. Muscle biopsy was obtained, and prednisone 1 mg/kg/day was initiated. Within 48 hours, her pain improved, and CK and transaminases began to decline. She was discharged on 60 mg prednisone daily.Post-discharge serologies showed positive anti–Jo-1, anti-PL7, and anti-SSA antibodies; ANA, anti–smooth muscle, anti-Mi-2, anti-SRP, anti-SSB, and anti–TIF1-γ were negative. SPEP showed hypoalbuminemia with mild alpha-2 elevation. Biopsy revealed perifascicular and perivascular inflammation without Mx1 expression, supporting antisynthetase or overlap myositis. She was started on azathioprine 50 mg daily. The combination of antibody profile, MRI findings, and biopsy confirmed antisynthetase syndrome (ASyS).
Discussion: This case demonstrates the diagnostic complexity of ASyS, a heterogeneous autoimmune myopathy with multisystem involvement that often mimics infectious or metabolic conditions. Severe rhabdomyolysis, fevers, transaminitis, diffuse myalgia, and an atypical rash initially broadened the differential, yet profound enzyme elevation and MRI abnormalities redirected evaluation toward autoimmune myositis. Myositis-specific antibodies and biopsy findings ultimately established the diagnosis. Early recognition is essential to prevent long-term morbidity.
Conclusions: Antisynthetase syndrome remains challenging to diagnose due to limited standardized criteria and its broad clinical spectrum. Timely identification is critical, as untreated cases carry substantial morbidity and mortality risk. Early immunosuppressive therapy improves outcomes, supports disease control, and guides appropriate malignancy screening, an important consideration given the syndrome’s associated cancer risk.